Overcoming the Challenges of Mixed Quantum-Classical Dynamics: From Theory to Drug Discovery Applications
Mixed quantum-classical (MQC) dynamics simulations are indispensable for modeling complex processes in photochemistry and drug discovery, yet their implementation faces significant theoretical and computational hurdles.
Overcoming the Challenges of Mixed Quantum-Classical Dynamics: From Theory to Drug Discovery Applications
Abstract
Mixed quantum-classical (MQC) dynamics simulations are indispensable for modeling complex processes in photochemistry and drug discovery, yet their implementation faces significant theoretical and computational hurdles. This article explores the foundational principles of MQC methods, their practical implementation using hybrid quantum-classical algorithms on near-term hardware, and the critical challenges of electronic decoherence and phase evolution. Through validation case studies and a comparative analysis of current approaches, we provide a roadmap for researchers and drug development professionals to optimize these simulations, highlighting their transformative potential for accelerating the design of novel therapeutics.
The Core Principles and Inherent Hurdles of Mixed Quantum-Classical Dynamics
Mixed Quantum-Classical (MQC) dynamics simulations are indispensable tools for modeling coupled electron-nuclear motion in molecular systems, a process fundamental to understanding photochemistry, charge transfer, and energy relaxation [1]. These methods treat electrons quantum mechanically while nuclei follow classical trajectories, making simulations of realistic molecular systems computationally feasible where fully quantum treatments are prohibitive [1] [2]. The core challenge in MQC implementations involves accurately capturing nonadiabatic effects—transitions between different electronic states—while maintaining proper electronic coherence and phase evolution [1] [3].
Within pharmaceutical research and drug development, MQC methods provide the foundation for simulating light-activated processes and predicting molecular interactions at an atomic level [4] [3]. This capability is transforming computational drug discovery by enabling more accurate prediction of protein-ligand binding, molecular stability, and toxicity profiles [5] [4].
Foundational MQC Methodologies and Equations
The theoretical framework for MQC dynamics originates from the Exact Factorization (XF) formalism, which provides a rigorous foundation for electron-nuclear dynamics by expressing the molecular wavefunction as a product of a nuclear wavefunction and a conditional electronic state [1]. This approach leads to coupled electronic and nuclear equations of motion:
Nuclear Equations: The exact classical force for a nuclear trajectory includes contributions from both the scalar potential and the time-dependent vector potential [1]:
𝐅ν = 𝐏˙ν(t) = -∇νϵ~ + A˙νElectronic Equations: The electronic time-dependent Schrödinger equation along nuclear trajectories incorporates both the Born-Oppenheimer Hamiltonian and electron-nuclear correlation operators [1]:
iℏ(d/dt)|ΦR¯¯(t)⟩ = (H^BO + H^ENC(1) + H^ENC(2) - ϵ~)|ΦR¯¯(t)⟩
Expansion of these equations in the Born-Oppenheimer basis yields multiple contributions to the time evolution of electronic coefficients [1]:
C˙j = C˙jEh + C˙jQM + C˙jPQM + C˙jDiv + C˙jPh
Table 1: Key Terms in Exact Factorization Formalism
| Term | Physical Significance | Role in MQC Dynamics |
|---|---|---|
| ϵ~(R¯¯,t) | Time-dependent scalar potential | Governs energy landscape for nuclear motion |
| Aν(R¯¯,t) | Time-dependent vector potential | Affects phase evolution of electronic states |
| H^ENC(1), H^ENC(2) | Electron-nuclear correlation operators | Describe beyond-Born-Oppenheimer effects |
| PQM Correction | Projected quantum momentum | Crucial for proper description of electronic coherence [1] |
| Phase Correction | Additional ℏ-order term | Governs proper phase evolution of BO coefficients [1] |
Frequently Asked Questions: MQC Implementation Challenges
Q1: Our surface hopping simulations show incorrect electronic state populations. What could be causing this?
Incorrect state populations often stem from inadequate treatment of electronic decoherence and improper phase evolution [1].
Troubleshooting Steps:
- Verify Decoherence Corrections: Ensure your implementation includes the Projected Quantum Momentum (PQM) correction, which naturally arises from the Exact Factorization framework and is essential for describing electronic coherence [1].
- Check Phase Evolution: Recent research identifies an additional phase-correction term of order ℏ that accompanies the PQM correction. The combined inclusion of both PQM and phase corrections provides a balanced description of decoherence and phase dynamics [1].
- Validate Against Benchmark Systems: Test your implementation on known model systems like the double-arch geometry (DAG) or two-dimensional nonseparable (2DNS) models to verify it reproduces characteristic nonadiabatic features such as Stückelberg oscillations [1].
Q2: How can we implement MQC dynamics using hybrid quantum-classical computing approaches?
Hybrid quantum-classical algorithms mitigate the limitations of current noisy quantum hardware while leveraging its potential for specific electronic structure calculations [2].
Implementation Protocol:
- Algorithm Selection: Employ the Variational Quantum Eigensolver (VQE) and Variational Quantum Deflation (VQD) algorithms to compute ground and excited state energies, gradients, nonadiabatic coupling vectors, and transition dipole moments [2].
- Software Integration: Implement by integrating molecular dynamics packages (like SHARC) with quantum computing frameworks (such as TEQUILA) [2].
- Dynamics Propagation: Use these quantum-computed electronic properties to propagate nonadiabatic molecular dynamics with established methods (e.g., Tully's fewest switches surface hopping) [2].
- Validation: Test the approach on well-characterized photochemical processes like cis-trans photoisomerization of methanimine and electronic relaxation of ethylene to verify qualitative accuracy against experimental and computational benchmarks [2].
Q3: What are the practical limitations of different electronic structure methods for excited-state MQC dynamics?
The choice of electronic structure method significantly impacts the accuracy and feasibility of MQC simulations [3].
Table 2: Electronic Structure Methods for Excited-State Dynamics
| Method | Strengths | Limitations | Best Use Cases |
|---|---|---|---|
| TD-DFT | Computationally efficient for large systems [3] | Accuracy depends on functional choice; challenges with charge-transfer states [3] | Initial screening of large molecular systems |
| Multiconfigurational Methods (CASSCF/CASPT2) | High accuracy for multireference problems [3] | Computationally expensive; active space selection critical [3] | Bond breaking, conical intersections, detailed photochemical studies |
| Hybrid QM/MM | Balances accuracy and computational cost [6] | QM/MM boundary effects can introduce artifacts [6] | Protein-ligand interactions in biological environments |
Q4: How do we properly simulate experimental observables like ultrafast spectra from MQC trajectories?
Connecting MQC simulations to experimental observables requires specialized post-processing [3]:
- Calculate Correlation Functions: Compute appropriate time-dependent correlation functions from your dynamics trajectories [3].
- Implement Wave Packet Propagation: For higher accuracy, especially in photochemical applications, use wave packet propagation methods rather than purely classical approaches [3].
- Model Experimental Conditions: Simulate specific spectroscopic techniques (e.g., pump-probe spectroscopy) by modeling the interaction with electric fields and calculating corresponding response functions [3].
Table 3: Essential Resources for MQC Simulations
| Resource Category | Specific Tools | Function/Purpose |
|---|---|---|
| Molecular Dynamics Packages | SHARC, Newton-X [3] | Propagate nuclear dynamics and manage electronic state transitions |
| Electronic Structure Codes | OpenMolcas, Psi4/pySCF [3] | Compute potential energy surfaces and nonadiabatic couplings |
| Quantum Computing Frameworks | TEQUILA [2] | Implement hybrid quantum-classical algorithms for electronic structure |
| Spectroscopy Tools | FCClasses3 [3] | Simulate vibrationally resolved electronic spectra |
| Benchmark Systems | Double-arch geometry (DAG), 2DNS models [1] | Validate new methodologies and implementations |
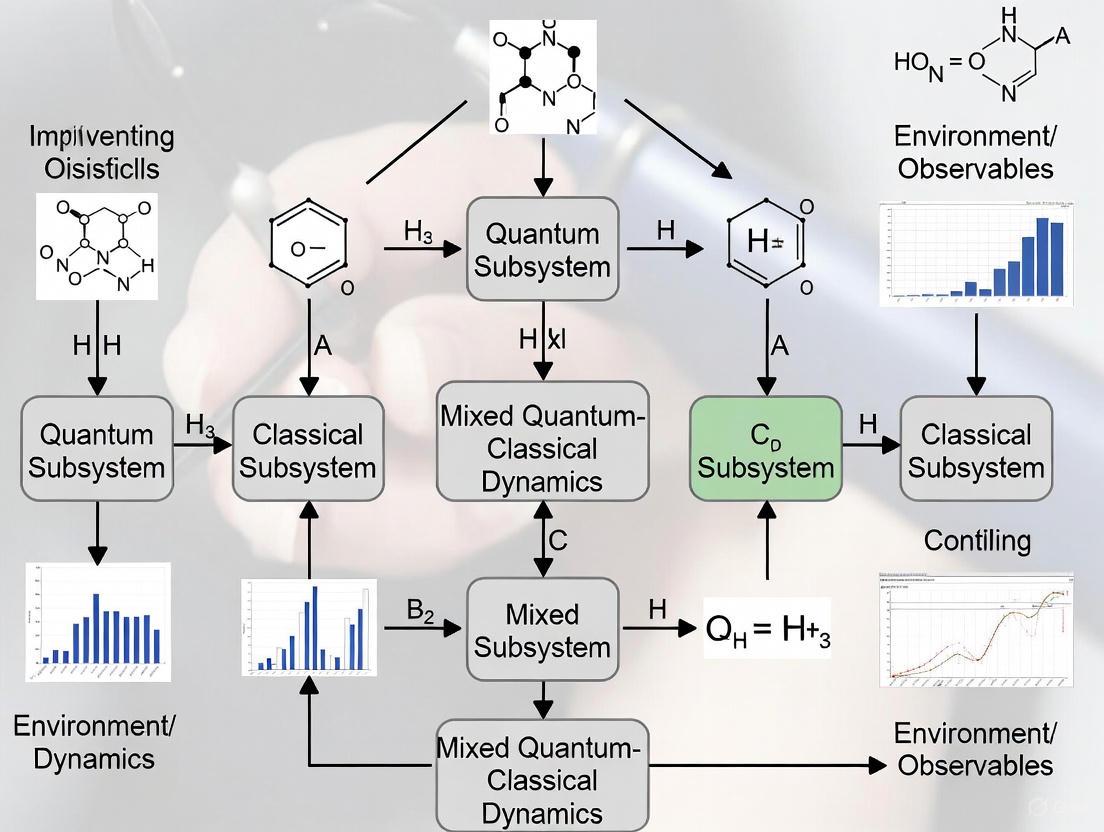
Workflow Visualization for MQC Implementation
MQC Simulation Workflow: This diagram outlines the fundamental iterative process of Mixed Quantum-Classical dynamics simulations, highlighting the feedback between quantum electronic and classical nuclear propagation.
Advanced Implementation: Quantum Computing Enhanced MQC
Hybrid Quantum-Classical Algorithm: This visualization shows the integration of quantum computing resources with classical MQC dynamics, particularly for computationally demanding electronic structure calculations.
Troubleshooting Guide: Frequently Asked Questions (FAQs)
FAQ 1: What are the primary symptoms of decoherence in my mixed quantum-classical dynamics simulation?
You can identify decoherence through several key symptoms in your simulation outputs. The most common is a loss of quantum information, where qubits that should be in superposition collapse to classical states, making the output of your quantum computation unreliable or meaningless [7]. You may also observe a breakdown of quantum interference patterns, which is essential for computations that rely on effects like those in a double-slit experiment [8]. Furthermore, a rapid decay in the fidelity of your computations, leading to incorrect results, is a direct indicator that coherence has been lost [7].
FAQ 2: My simulation results show incorrect phase relationships. Could this be linked to the "phase evolution problem"?
Yes, incorrect phase relationships are a hallmark of the phase evolution problem. In mixed quantum-classical dynamics, a lack of proper phase-correction within the equations of motion can lead to an inaccurate account of electronic phase evolution [9]. This results in flawed nuclear forces and a failure to capture key nonadiabatic features in the system. Advanced frameworks, such as the Exact Factorization approach, are being developed to unify the treatment of decoherence and phase evolution by incorporating these missing phase-correction terms [9].
FAQ 3: What are the most effective strategies to mitigate decoherence in near-term quantum simulations?
A multi-layered approach is required to mitigate decoherence. The table below summarizes the primary strategies:
| Mitigation Strategy | Brief Description | Key Function |
|---|---|---|
| Quantum Error Correction (QEC) [7] | Uses quantum codes (e.g., surface codes) to detect and correct errors without direct measurement. | Protects logical qubit information by encoding it across multiple physical qubits. |
| Environmental Isolation [7] | Uses vacuum chambers and shielding to protect the quantum processor. | Minimizes unwanted interactions with the external environment that cause decoherence. |
| Cryogenic Cooling [7] | Cools quantum systems (e.g., superconducting qubits) to millikelvin temperatures. | Reduces thermal noise and fluctuations that disrupt quantum states. |
| Material Engineering [7] | Designs cleaner substrates and interfaces for qubit fabrication. | Reduces intrinsic material noise sources, improving coherence times. |
| Decoherence-Free Subspaces (DFS) [7] | Encodes quantum information in special configurations immune to certain common noise sources. | Provides a inherent resilience to specific types of environmental decoherence. |
FAQ 4: How does the choice of qubit technology impact susceptibility to decoherence and phase errors?
Different qubit platforms offer varying trade-offs between coherence time, operational speed, and inherent noise resistance, which directly impacts their susceptibility to these problems [7].
- Superconducting Qubits: Offer fast gate speeds but are sensitive to noise and have relatively short coherence times, requiring sophisticated error mitigation [7] [10].
- Trapped Ion Qubits: Have significantly longer coherence times and high-fidelity operations, but their gate speeds are generally slower [7] [10].
- Photonic Qubits: Are naturally resistant to decoherence because photons interact very little with their environment. This makes them promising for quantum communication, but creating the necessary interactions for universal quantum computation is challenging [7].
FAQ 5: Are there new theoretical developments that simultaneously address decoherence and phase evolution?
Yes, recent research has made progress in unifying these challenges. New mixed quantum-classical equations of motion derived from the Exact Factorization framework have been proposed. These formulations introduce not only a projected quantum momentum correction but also a crucial phase-correction term. This provides a unified and more rigorous account of both electronic coherence and phase evolution, and their combined effect on nuclear forces [9].
Experimental Protocols & Methodologies
Protocol: Implementing a Mixed Quantum-Classical Dynamics Workflow using Hybrid Hardware
This protocol outlines the steps for running nonadiabatic molecular dynamics by computing electronic properties on quantum hardware, a method demonstrated in the integration of the SHARC molecular dynamics package with the TEQUILA quantum computing framework [2].
Objective: To propagate nonadiabatic molecular dynamics using the Tully's fewest switches surface hopping method, with essential electronic properties computed on a quantum processor.
Key Research Reagent Solutions:
| Item | Function in the Experiment |
|---|---|
| SHARC Program Package | A classical molecular dynamics program used to propagate nuclear motion and manage the surface hopping algorithm [2]. |
| TEQUILA Framework | A quantum programming framework used to formulate and execute quantum circuits on simulators or hardware [2]. |
| Variational Quantum Eigensolver (VQE) | A hybrid quantum-classical algorithm used to compute the ground-state energy of molecules on near-term quantum devices [2] [11]. |
| Variational Quantum Deflation (VQD) | An algorithm used to compute excited state energies, which are essential for nonadiabatic transitions [2]. |
| Tully's Fewest Switches Surface Hopping | The mixed quantum-classical dynamics method that describes nonadiabatic transitions between electronic states [2]. |
Methodology:
- System Initialization: Define the molecular system and initialize the nuclear coordinates and momenta on a chosen electronic potential energy surface.
- Quantum Computation of Electronic Properties: For the current nuclear configuration, use the TEQUILA framework to run VQE and VQD algorithms on a quantum processor to calculate:
- Ground and excited state energies
- Energy gradients (forces)
- Nonadiabatic coupling vectors
- Transition dipole moments
- Classical Nuclear Propagation: The SHARC package uses the computed forces to classically propagate the nuclear coordinates for a short time step.
- Surface Hopping Decision: Based on the nonadiabatic couplings and quantum amplitudes, stochastically determine if a hop to a different electronic state occurs.
- Iteration: Repeat steps 2-4 for the duration of the simulation, continuously using the quantum computer to update the electronic structure properties.
- Validation: Benchmark the resulting dynamics against experimental data or high-level classical computational studies, for example, by studying photoisomerization processes like in methanimine or ethylene [2].
The following workflow diagram illustrates the interaction between the classical and quantum computing components in this protocol:
Visualizing the Decoherence Process
To effectively troubleshoot decoherence, it is crucial to understand its fundamental mechanism: environmentally-induced superselection, or einselection [8]. The diagram below illustrates how a quantum system loses its coherence through interaction with a large environment.
The performance of different quantum systems and error correction methods can be quantitatively assessed. The table below summarizes key metrics relevant to managing decoherence and phase errors.
Table 1: Comparative Analysis of Decoherence Metrics and Mitigation Performance
| Qubit Technology / Method | Typical Coherence Time | Operational Speed | Primary Error Source | Mitigation Overhead (Physical Qubits per Logical Qubit) |
|---|---|---|---|---|
| Superconducting Qubits [7] [10] | Microseconds to Milliseconds | Fast (ns gate speeds) | Thermal noise, Imperfect isolation | High (Dozens to hundreds for QEC) |
| Trapped Ion Qubits [7] [10] | Longer (up to seconds) | Slower (ms gate speeds) | Spontaneous emission, Laser phase noise | High (Dozens to hundreds for QEC) |
| Quantum Error Correction (QEC) [7] | Extends effective time | Slower due to encoding | All of the above | Very High (Required for fault-tolerance) |
| Decoherence-Free Subspaces (DFS) [7] | N/A (Inherent resistance) | Platform-dependent | Specific collective noise | Low (Relies on specific encoding) |
Frequently Asked Questions (FAQs)
Q1: What is the primary numerical challenge when implementing exact numerical propagation of the XF equations? The primary challenge is the emergence of severe numerical instabilities, particularly in regions where the nuclear probability density is low. This instability is largely driven by the quantum momentum term, defined as ( r(y,t) = \frac{\nabla_y |\psi(y,t)|}{|\psi(y,t)|} ). Since this term involves division by the nuclear wavefunction amplitude, it becomes singular or near-singular as ( |\psi(y,t)| ) approaches zero, leading to divergent behavior in the calculations [12].
Q2: How does the nuclear wavefunction's behavior contribute to this instability? As a nuclear wavepacket evolves and bifurcates (e.g., during photodissociation or passing through a region of strong non-adiabatic coupling), spatial separation creates nodes and low-density regions. The XF formalism's coupling terms are highly sensitive to this separation. The resulting near-discontinuous steps in the time-dependent potential energy surface (TDPES) further exacerbate numerical difficulties, regardless of the strength of the non-adiabatic coupling itself [12].
Q3: My XF-based mixed quantum-classical (MQC) simulation is unstable. What are the main strategy differences I should consider? A key strategic difference lies in how the crucial electron-nuclear coupling (specifically, the quantum momentum) is evaluated. You can choose between algorithms that use coupled trajectories versus those that use auxiliary trajectories to approximate this term [13] [14].
- Coupled Trajectory (CT) Methods: These include approaches like the Coupled-Trajectory Mixed Quantum-Classical (CTMQC) and Coupled-Trajectory Surface Hopping (CTSH) methods. In these, ensembles of trajectories are evolved concurrently and "talk" to each other to compute the quantum momentum based on the collective nuclear density [13].
- Auxiliary Trajectory Methods: These include methods like SHXF. Here, the primary trajectories are supplemented with auxiliary trajectories designed to mimic the delocalization of the nuclear density and estimate the coupling terms without direct inter-trajectory coupling [13].
Q4: Beyond numerical stability, what physical accuracy conditions should a robust XF-based MQC method satisfy? When developing or selecting an XF-based method, verify that it respects two key exact conditions [14]:
- Zero Population Transfer Away from Coupling Regions: Electronic population transfer should only occur in spatial regions where non-adiabatic couplings are significant.
- Conservation of Total Energy: The method must conserve the total energy of the isolated molecular system.
Q5: Are there new corrections derived from XF that improve the description of electronic coherence and phase evolution? Yes, recent derivations have uncovered two important correction terms of order ( \hbar ) that act on the electronic subsystem [1]:
- The Projected Quantum Momentum (PQM) Correction: Directly responsible for inducing electronic decoherence.
- A Phase Correction: Governs the proper phase evolution of the Born-Oppenheimer coefficients. The combined inclusion of both terms in the equations of motion provides a unified and rigorous description of both decoherence and phase dynamics, leading to more accurate simulation of features like Stückelberg oscillations [1].
Troubleshooting Guides
Issue 1: Numerical Instabilities in Low-Density Regions
Symptoms
- Simulations crash or produce non-physical results (e.g., NaN errors) when nuclear wavepackets split or move into classically forbidden regions.
- Observed spikes in the time-dependent potentials or forces.
Diagnostic Steps
- Monitor the Nuclear Density: Track the value of ( |\psi(R,t)|^2 ) along your nuclear coordinates. Instabilities are likely to originate in regions where this value drops below a certain threshold [12].
- Analyze the Quantum Momentum: Plot the quantum momentum term ( \frac{\nabla_R |\psi|}{|\psi|} ). Check for abnormally large values that indicate the onset of divergence [12].
Solutions
- Switch to a Trajectory-Based Framework: Consider moving from a full quantum nuclear treatment to an MQC scheme that uses coupled or auxiliary trajectories. This avoids direct computation with a low-amplitude wavefunction [13] [12].
- Investigate Different Quantum Momentum Computations: If using a trajectory method, test the stability of different quantum momentum evaluation schemes (coupled vs. auxiliary) for your specific system [13] [14].
- Employ a Regularization Technique: Develop or implement a mathematical regularization that places a lower bound on the nuclear density in the denominator of the quantum momentum term, preventing division by zero. (Note: This must be done with care to preserve physical accuracy).
Issue 2: Inaccurate Electronic Dynamics and Decoherence
Symptoms
- Electronic populations or coherences do not agree with benchmark quantum dynamics results.
- Missing characteristic quantum effects like Stückelberg oscillations.
Diagnostic Steps
- Check for PQM and Phase Corrections: Verify the equations of motion in your chosen algorithm. Many standard MQC methods lack the rigorous PQM and phase corrections derived from XF [1].
- Benchmark on Simple Models: Test your implementation on well-known model systems (e.g., simple avoided crossing, double-arch geometry) where exact quantum results are available for comparison [1].
Solutions
- Implement the PQM and Phase Corrections: Incorporate the derived terms from the XF framework into your electronic equation of motion. The correction to the time evolution of BO coefficients ( Cj ) is [1]: ( \dot{C}j = \dot{C}j^{\text{Eh}} + \dot{C}j^{\mathrm{QM}} + \dot{C}j^{\mathrm{PQM}} + \dot{C}j^{\mathrm{Div}} + \dot{C}j^{\mathrm{Ph}} ) Ensure both ( \dot{C}j^{\mathrm{PQM}} ) (decoherence) and ( \dot{C}_j^{\mathrm{Ph}} ) (phase evolution) are included.
- Validate Against Exact Calculations: Use published results for the TDPES and vector potentials from exact XF calculations on small systems (e.g., 1D ( \text{H}_2^+ ) models) to calibrate your approximations [13].
Experimental Protocols & Methodologies
Protocol 1: Implementing a Coupled-Trajectory XF Scheme (CTMQC/CTSH)
This protocol outlines the steps for running a simulation using the coupled-trajectory approach to compute the quantum momentum [13].
Objective: To propagate non-adiabatic dynamics using an ensemble of classical nuclear trajectories whose evolution is coupled through the XF-derived quantum momentum.
Workflow Overview:
Step-by-Step Instructions:
- Initialization:
- Generate an ensemble of ( N ) independent classical trajectories ( { \mathbf{R}^{(I)}(t), \mathbf{P}^{(I)}(t) } ), where ( I = 1, ..., N ). Sample initial nuclear positions ( \mathbf{R}^{(I)}(0) ) and momenta ( \mathbf{P}^{(I)}(0) ) from the initial wavepacket Wigner distribution [13].
- Assign each trajectory an initial electronic wavefunction ( |\Phi^{(I)}(0)\rangle ).
Electronic Propagation:
- For each trajectory ( I ) at time ( t ), compute the Hamiltonian in the Born-Oppenheimer basis (energies ( \epsilonj ), non-adiabatic coupling vectors ( \mathbf{d}{jk} )).
- Propagate the electronic coefficients ( Cj^{(I)}(t) ) according to the expanded equation of motion that includes the quantum momentum (QM) term [13] [1]: [ i\hbar \dot{C}j^{(I)} = \epsilonj Cj^{(I)} - i\hbar \sumk \frac{\mathbf{P}^{(I)}}{M} \cdot \mathbf{d}{jk} Ck^{(I)} - i\hbar \sumk \frac{\bm{\mathcal{P}}^{(I)}}{2M} \cdot \mathbf{D}{jk} Ck^{(I)} + \text{(other corrections)} ] Here, ( \bm{\mathcal{P}}^{(I)} ) is the quantum momentum for trajectory ( I ).
Quantum Momentum Calculation:
- The key step: compute the quantum momentum ( \bm{\mathcal{P}}^{(I)} ) for each trajectory. This is not derived from its own wavefunction but from the entire ensemble.
- In CTMQC, this is often approximated as [13]: [ \bm{\mathcal{P}}(\mathbf{R}^{(I)}(t), t) \approx \frac{\sum{J=1}^N \nabla \sigma(\mathbf{R}^{(I)}(t) - \mathbf{R}^{(J)}(t))}{\sum{J=1}^N \sigma(\mathbf{R}^{(I)}(t) - \mathbf{R}^{(J)}(t))} ] where ( \sigma ) is a smoothing function (e.g., a Gaussian). This couples the trajectories.
Nuclear Force Calculation and Propagation:
- Compute the force on each nucleus in trajectory ( I ). In the exact XF, the force contains terms from the TDPES ( \epsilon ) and the vector potential ( \mathbf{A} ) [1]: [ \mathbf{F}\nu = -\nabla\nu \tilde{\epsilon} + \dot{\mathbf{A}}_\nu ]
- In MQC approximations, this is translated into a force that includes a component from the quantum momentum. Propagate the nuclear positions and momenta of all trajectories using a suitable classical integrator (e.g., Velocity Verlet).
Loop and Analysis:
- Repeat steps 2-4 until the total simulation time is reached.
- Analyze ensemble properties (e.g., average population, spatial distribution).
Protocol 2: Computing the Exact Quantum Momentum from a Nuclear Wavefunction
For methods that treat the nuclei fully quantum mechanically, or for benchmarking trajectory-based methods, this protocol details the calculation of the quantum momentum [12].
Objective: To accurately compute the quantum momentum term ( \mathbf{r}(R,t) = \frac{\nabla_R |\psi(R,t)|}{|\psi(R,t)|} ) from a discretized nuclear wavefunction ( \psi(R,t) ).
Workflow Overview:
Step-by-Step Instructions:
- Represent the Wavefunction: Ensure the full molecular wavefunction ( \Psi(r, R, t) ) or the factored nuclear wavefunction ( \psi(R, t) ) is represented on a numerical grid in the nuclear coordinate ( R ).
Compute the Nuclear Density: Calculate the marginal nuclear probability density ( \rho(R, t) = |\psi(R, t)|^2 ) at every grid point.
Calculate the Gradient: Compute the spatial gradient of the density, ( \nabla_R \rho(R, t) ), using a stable numerical method (e.g., a centered finite difference scheme).
Form the Quantum Momentum: The quantum momentum is given by: [ \mathbf{r}(R,t) = \frac{\nablaR |\psi(R,t)|}{|\psi(R,t)|} = \frac{\nablaR \rho(R,t)}{2 \rho(R,t)} ] This identity is used for numerical stability as it avoids explicitly taking the square root of the density.
Handle Low-Density Regions:
- This is the most critical step. Identify grid points where ( \rho(R, t) ) falls below a pre-defined, small threshold (e.g., ( 10^{-8} )).
- For these points, do not compute the ratio. Instead, implement a regularization strategy. Options include:
- Setting ( \mathbf{r}(R,t) ) to zero at these points.
- Using a smooth cutoff function that tapers the quantum momentum to zero as density decreases.
- The choice of strategy can significantly impact stability and accuracy and should be tested and documented.
The Scientist's Toolkit: Research Reagent Solutions
The following table catalogs the essential "research reagents" – the computational methods, model systems, and mathematical terms – central to working with the Exact Factorization formalism.
Table 1: Key Reagents in XF Methodology Development
| Reagent Name | Type | Primary Function | Key Considerations |
|---|---|---|---|
| Quantum Momentum (( \mathcal{P} )) [12] | Mathematical Term | Encodes the effect of nuclear density delocalization on electronic evolution; primary driver of decoherence in XF-MQC. | Source of numerical instability in low-density regions. Must be approximated in trajectory methods. |
| Coupled Trajectories [13] [14] | Algorithmic Approach | Enables calculation of the quantum momentum by having an ensemble of trajectories exchange information. | Computationally expensive; can be sensitive to the number of trajectories and the smoothing kernel used. |
| Auxiliary Trajectories [13] [14] | Algorithmic Approach | Estimates the quantum momentum for a primary trajectory using nearby "auxiliary" trajectories, avoiding direct coupling. | Can be less accurate than fully coupled methods but may offer improved numerical stability. |
| Time-Dependent Potential Energy Surface (TDPES, ( \epsilon )) [13] | Mathematical Term / Observable | The exact scalar potential driving the nuclear motion in the XF picture. Contains steps and gauge-dependent features that signal non-adiabatic events. | Invaluable for interpreting dynamics but requires an exact solution of the TDSE for a molecule for benchmarking. |
| Projected Quantum Momentum (PQM) & Phase Corrections [1] | Mathematical Term / Correction | Rigorously derived terms that ensure accurate electronic decoherence and phase evolution in MQC simulations. | Their combined inclusion is necessary for reproducing effects like Stückelberg oscillations. |
| Model Systems (e.g., 1D H₂⁺, Tully Models) [13] [12] | Benchmarking Tool | Simple, computationally tractable systems for which exact quantum and exact XF results can be computed to test new approximations. | Essential for the initial development and validation of any new XF-based method or code. |
Method Comparison and Selection Guide
Table 2: Comparison of XF-Based Mixed Quantum-Classical Methods
| Method | Quantum Momentum Evaluation | Key Features | Best For | Stability & Accuracy Notes |
|---|---|---|---|---|
| CTMQC [13] | Coupled Trajectories | Derived directly from XF; includes QM correction in nuclear force. | Systems with strong non-adiabatic coupling and clear wavepacket splitting. | More stable than exact propagation; respects zero transfer condition with modified QM [13] [14]. |
| CTSH [13] | Coupled Trajectories | QM term incorporated into a surface-hopping framework. | Researchers familiar with surface hopping who want added decoherence. | Similar stability to CTMQC; performance depends on how QM affects hops [13]. |
| SHXF [13] | Auxiliary Trajectories | Uses auxiliary trajectories to mimic nuclear density for QM estimation. | Larger systems where full trajectory coupling is computationally prohibitive. | Generally stable, but accuracy depends on the quality of the auxiliary trajectory approximation [13]. |
| FENDy [12] | From Nuclear Wavefunction | Uses a complex potential instead of a vector potential in the nuclear equation. | Benchmarking and fundamental studies where exact nuclear quantum effects are critical. | Prone to the same numerical instabilities in low-density regions as other exact methods [12]. |
Frequently Asked Questions
FAQ 1: What makes on-the-fly electronic structure calculations so computationally demanding in nonadiabatic dynamics?
The primary cost arises from the need to compute a set of expensive electronic properties—including energies, forces, and nonadiabatic coupling vectors between electronic states—at each time step for every classical nuclear trajectory. These properties must be calculated using quantum mechanical (QM) methods, which scale poorly with system size. For instance, a simulation of a medium-sized molecule can require hundreds of thousands of CPU hours [15]. Furthermore, for a system involving N_s electronic energy surfaces, the number of nonadiabatic coupling vectors that must, in principle, be computed scales with N_s (N_s - 1)/2, creating a major bottleneck [16].
FAQ 2: Why can't we pre-compute these properties to save time? Pre-computing potential energy surfaces (PESs) for excited-state dynamics is often infeasible due to the curse of dimensionality. The number of points needed to map a PES grows exponentially with the number of degrees of freedom (atomic coordinates). For a relatively small 33-dimensional model, generating a reference data set with the same point density as a complete 1-dimensional model would require an impossible 3.45 × 10^69 grid points [15]. On-the-fly strategies avoid this by calculating energies and couplings only at the specific geometries visited by the trajectories during the simulation [15].
FAQ 3: How does the choice of electronic structure method impact computational cost? The trade-off between accuracy and cost is severe. While Density Functional Theory (DFT) is widely used, its accuracy is not always sufficient [17]. More accurate methods, like the coupled-cluster theory (CCSD(T)), are considered the "gold standard" but have terrible scaling; doubling the number of electrons in the system makes the computations 100 times more expensive [17]. Multi-reference configuration interaction (MRCI) methods are also highly accurate but computationally demanding, especially for larger configuration spaces [16].
FAQ 4: Are there specific electronic properties that are particularly expensive to compute? Yes, nonadiabatic coupling vectors are a significant bottleneck. These couplings are essential for describing how energy is transferred between electronic states near conical intersections, but their computation is "computationally demanding" [16]. They also feature "sharp, narrow spikes" around certain nuclear geometries, which necessitates very small time steps during dynamics integration, further increasing the number of required QM calculations [15].
FAQ 5: What role does system size play in the computational cost?
Computational cost scales poorly with system size. Traditional DFT calculations, for example, involve repeated matrix diagonalizations where the computational cost scales as O(N³) with the number of electrons or basis functions N [18]. This cubic scaling means that simulating large or complex materials quickly becomes "extremely resource-consuming" [18].
Table 1: Key Computational Bottlenecks and Their Impact
| Bottleneck | Computational Cost/Scalability | Primary Impact |
|---|---|---|
| Property Calculation per Time Step [15] | Hundreds of thousands of CPU hours for a medium-sized molecule | Makes long-time-scale or large-system simulations prohibitively expensive |
| Electronic Structure Method [17] | CCSD(T) cost scales ~100x for 2x electrons; DFT has O(N³) scaling [18] | Forces a trade-off between accuracy and feasibility for large systems |
| Number of Coupling Vectors [16] | Scales as Ns (Ns - 1)/2 for N_s electronic states | Simulations with many coupled states become intractably slow |
| Mapping Full Potential Energy Surfaces [15] | Points needed grow exponentially with system dimensionality (e.g., 3.45×10^69 for a 33D model) | Makes on-the-fly calculation the only viable approach for most systems |
Experimental Protocols for Mitigating Bottlenecks
Protocol 1: Hybrid Quantum-Classical Computing for Molecular Dynamics This protocol uses a hybrid quantum-classical algorithm to offload the expensive electronic structure calculations to a quantum computer, potentially achieving a "quantum advantage" for chemical simulations [2].
- Software Integration: Use an integrated framework like the SHARC molecular dynamics package coupled with the TEQUILA quantum computing platform [2].
- Electronic Property Calculation: For each geometry visited by the classical nuclear trajectory, employ variational quantum algorithms (e.g., Variational Quantum Eigensolver) on the quantum processor to compute ground and excited state energies, gradients, and nonadiabatic coupling vectors [2].
- Classical Propagation: Feed the computed electronic properties into a classical computer to propagate the nuclear dynamics using a method like Tully's surface hopping [2].
- Validation: Validate the approach by studying photochemical processes such as the cis-trans photoisomerization of methanimine and the electronic relaxation of ethylene [2].
Protocol 2: Machine Learning (ML) for Accelerated Nonadiabatic Dynamics This protocol uses machine learning to create surrogate models that predict electronic properties, drastically reducing the number of expensive QM calculations required [15].
- Generate Training Data: Perform a limited number of accurate QM calculations (e.g., 10,000 points or fewer) for a representative set of molecular geometries to create a training dataset. This should include energies, forces, and nonadiabatic couplings [15].
- Train ML Model: Train a machine learning model (e.g., using Kernel Ridge Regression) on the generated dataset to learn the mapping from nuclear coordinates to electronic properties [15].
- Run ML-Driven Dynamics: Run the nonadiabatic dynamics (e.g., surface hopping) using the ML-predicted properties for most time steps.
- Handle Critical Regions: Implement a safeguard: when the ML-predicted energy gap between states falls below a critical threshold (e.g., 0.03 hartree), switch to an explicit QM calculation for that step to ensure accuracy near conical intersections where nonadiabatic couplings spike [15].
Protocol 3: Algorithmic Optimizations in Mixed Quantum-Classical Dynamics This protocol involves using algorithmic approximations within the dynamics simulation to reduce cost while maintaining acceptable accuracy [16].
- Method Selection: Choose the appropriate approximation:
- Time-Derivative Couplings: Use the Hammes-Schiffer-Tully (HST) model, which computes nonadiabatic couplings via wavefunction overlaps, instead of the more expensive nonadiabatic coupling vectors. This is faster for systems with small multiconfiguration spaces [16].
- Partially Coupled Equations: Neglect nonadiabatic coupling vectors between specific pairs of states that are known to be weakly interacting. This can speed up simulations with deviations in population below 5% [16].
- Benchmarking: Compare the results and computational cost of the approximate simulation against a benchmark dynamics calculation that uses the full set of nonadiabatic coupling vectors to validate the approach for the system of interest [16].
Workflow and Property Dependencies
The diagram below illustrates the core computational workflow of on-the-fly mixed quantum-classical dynamics and the critical dependencies between the calculated electronic properties.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools and Methods
| Tool / Method | Function | Relevant Context |
|---|---|---|
| SHARC & TEQUILA [2] | Integrated framework for mixed quantum-classical dynamics on quantum computers. | Replaces classical electronic structure calculations with hybrid quantum-classical algorithms. |
| Multi-task MEHnet [17] | A deep learning model trained with CCSD(T) data to predict multiple electronic properties from a single model. | Achieves high accuracy without the cost of repeated CCSD(T) calculations. |
| Decoherence-Corrected FSSH [15] | A popular mixed quantum-classical dynamics method for simulating nonadiabatic processes. | The target method for which ML potentials are often developed. |
| Kernel Ridge Regression (KRR) [15] | A machine learning algorithm used to create potentials for excited-state energies and forces. | Used to learn the mapping from nuclear coordinates to electronic properties. |
| Exact Factorization (XF) [1] | A rigorous theoretical framework for deriving improved mixed quantum-classical equations of motion. | Provides a foundation for more accurate treatment of decoherence and phase evolution. |
| NextHAM Deep Learning Model [18] | A neural network for predicting electronic-structure Hamiltonians, bypassing the self-consistent loop of DFT. | Delivers DFT-level precision with dramatically improved computational efficiency. |
FAQ: Troubleshooting Common MQC Implementation Challenges
1. My Ehrenfest dynamics simulations show incorrect long-term energy distributions and detail balance. What is the cause and how can I address it?
This is a known fundamental limitation of the Mean-Field Ehrenfest (MFE) method. MFE causes the classical bath to evolve on an average potential surface, which prevents proper wave-packet branching and can lead to unphysical heating to an infinite temperature over long timescales [19]. To address this:
- Consider Alternative Methods: Switch to a surface-hopping method like the mapping approach to surface hopping (MASH) or Tully's fewest switches surface hopping (FSSH). These methods stochastically select a single potential surface for the classical bath to evolve on, which better captures wave-packet branching and, in the case of MASH, can obey detail balance [19].
- Consult the Protocol: Refer to the provided experimental protocol for MFE to ensure correct implementation and understand its inherent limitations for your specific observable [19].
2. When implementing surface hopping, how should I handle frustrated hops and ensure energy conservation?
Frustrated hops occur when a trajectory attempts to hop to another electronic state but lacks the necessary nuclear kinetic energy. The standard procedure is to disallow the hop; the trajectory continues on its current electronic state without a velocity adjustment. To ensure total energy conservation, successful hops typically require a rescaling of the nuclear momenta in the direction of the nonadiabatic coupling vector [20].
3. What steps can I take to improve the description of electronic decoherence and phase evolution in my MQC simulations?
The Exact Factorization (XF) framework provides a rigorous foundation for deriving corrections that address these issues. Recent work shows that including both a Projected Quantum Momentum (PQM) correction and a previously unidentified phase-correction term, both of order ℏ, is essential [1].
- Implement XF-derived corrections: Incorporate the PQM and phase-correction terms into your equations of motion. Benchmark tests on model systems demonstrate that this combined approach accurately captures key nonadiabatic features like electronic coherence and proper phase evolution [1].
- Validate with Model Systems: Test your implementation on standard models like the double-arch geometry (DAG) or two-dimensional nonseparable (2DNS) systems to verify the performance of these corrections [1].
4. My quantum chemistry calculations for on-the-fly MQC dynamics are computationally prohibitive. Are there emerging strategies to reduce this cost?
Yes, hybrid quantum-classical algorithms that leverage quantum computing are emerging as a promising strategy. One approach integrates the classical molecular dynamics package SHARC with the quantum computing framework TEQUILA [2].
- Utilize Hybrid Quantum-Classical Hardware: This framework uses algorithms like the Variational Quantum Eigensolver (VQE) to compute the required electronic properties (energies, gradients, nonadiabatic couplings) on a quantum processor, while the nuclear trajectories are propagated classically. This can significantly mitigate the classical computational demand of the quantum chemistry part [2].
- Application: This method has been successfully validated for nonadiabatic simulations in molecules like methanimine and ethylene [2].
Experimental Protocols for Key MQC Methods
Protocol 1: Mean-Field Ehrenfest (MFE) Dynamics for Polaron Spectral Functions
This protocol outlines the steps for applying MFE to calculate the spectral function of the one-dimensional Holstein polaron [19].
System Initialization:
- Model: Set up the one-dimensional Holstein Hamiltonian with periodic boundary conditions.
- Parameters: Define the electronic hopping amplitude (
J), electron-phonon coupling strength (g), and phonon frequency (ω0). - Initial Conditions: Sample initial classical nuclear coordinates and momenta (representing phonons) from an appropriate thermal distribution (e.g., Wigner distribution). Initialize the electronic wavefunction, typically starting with a single site excitation.
Dynamics Propagation:
- Electronic Propagation: At each time step, solve the time-dependent Schrödinger equation for the electronic coefficients ( Cj(t) ): ( i\hbar \dot{C}j = \epsilonj Cj - \sum{\nu,k} \frac{P\nu}{M\nu} \cdot \textbf{d}{\nu,jk} Ck ) where ( \textbf{d}{\nu,jk} ) is the nonadiabatic coupling vector.
- Nuclear Propagation: Evolve the classical nuclei using forces computed from the average Ehrenfest potential: ( \mathbf{F}\nu = -\nabla\nu \langle \Phi{\mathbf{R}}(t) | \hat{H}{BO} | \Phi_{\mathbf{R}}(t) \rangle )
- Numerical Integration: Use a suitable numerical integrator (e.g., Velocity Verlet) to propagate the coupled electronic and classical equations for a sufficiently long time.
Spectral Function Calculation:
- Correlation Function: Compute the time-dependent correlation function ( C(t) = \langle c(t) c^\dagger(0) \rangle ) from the simulation data.
- Fourier Transform: Calculate the spectral function ( A(\omega) ) as the Fourier transform of ( C(t) ).
Protocol 2: Exact Factorization with PQM and Phase Corrections
This protocol describes implementing the XF-based MQC method with corrections for decoherence and phase evolution [1].
Exact Factorization Setup:
- Express the total molecular wavefunction as a product of a nuclear wavefunction and a conditional electronic wavefunction, ( |\Psi(\mathbf{R},t)\rangle = \chi(\mathbf{R},t) |\Phi_{\mathbf{R}}(t)\rangle ), with the partial normalization condition.
Equation of Motion Derivation:
- Derive the coupled equations for the nuclear and electronic components from the time-dependent Schrödinger equation.
- Identify the key terms: the time-dependent vector potential ( \mathbf{A}\nu ), scalar potential ( \tilde{\epsilon} ), and the electron-nuclear correlation operators ( \hat{H}^{(1)}{\mathrm{ENC}} ) and ( \hat{H}^{(2)}_{\mathrm{ENC}} ).
Inclusion of Corrections:
- PQM Correction: Include the projected quantum momentum term ( \dot{C}_j^{\mathrm{PQM}} ) in the electronic equation.
- Phase Correction: Include the additional phase-correction term ( \dot{C}_j^{\mathrm{Ph}} ) of order ( \hbar ) that governs the proper phase evolution of the Born-Oppenheimer coefficients.
Benchmarking:
- Validate the implementation on benchmark systems like the double-arch geometry (DAG) and two-dimensional nonseparable (2DNS) models to confirm accurate reproduction of Stückelberg oscillations and nuclear-density distributions [1].
Comparative Analysis of MQC Methods
The table below summarizes the key characteristics, strengths, and weaknesses of several common MQC methods.
Table 1: Comparison of Common Mixed Quantum-Classical Methods
| Method | Key Principle | Strengths | Weaknesses / Common Issues |
|---|---|---|---|
| Mean-Field Ehrenfest (MFE) | Classical nuclei evolve on a single, average potential energy surface created by the quantum electrons [19]. | - Simple implementation [19]- Computationally efficient [19] | - Lacks wave-packet branching [19]- Can violate detail balance, leading to unphysical energy distributions over time [19] |
| Fewest Switches Surface Hopping (FSSH) | Classical nuclei evolve on a single potential surface; stochastic hops between surfaces occur based on quantum probabilities [20]. | - Captures wave-packet branching [19]- Widely used and tested [20] | - Requires decoherence corrections- Frustrated hops can be an issue [20] |
| Mapping Approach to Surface Hopping (MASH) | Classical nuclei evolve on a potential surface selected deterministically from mapping variables [19]. | - Obeys detail balance [19]- Provides accurate population dynamics [19] | - Newer method, broader applicability still under investigation [19] |
| Exact Factorization (XF) with PQM & Phase Corrections | Derives equations from a rigorous factorization of the molecular wavefunction [1]. | - Provides a unified, rigorous account of electronic coherence and phase evolution [1]- Corrections arise naturally from the formalism [1] | - Higher computational complexity [1]- Requires implementation of additional correction terms [1] |
Method Workflows and Logical Relationships
Diagram 1: MQC Dynamics Workflow Comparison. This diagram contrasts the fundamental procedural steps for Ehrenfest and Surface Hopping methods.
Diagram 2: Logical Relationships of MQC Methodologies. This diagram shows how different MQC methods conceptually relate to the core challenge of electron-nuclear correlation and how the Exact Factorization framework unifies and improves upon them.
Table 2: Key Computational Tools and Algorithms for MQC Research
| Item / Algorithm | Function / Purpose | Example Application / Note |
|---|---|---|
| Holstein Model Hamiltonian | A simple, standard model for testing MQC methods that describes a single electronic carrier interacting with local phonon modes on a lattice [19]. | Benchmarking method performance for polaron formation and dynamics [19]. |
| Variational Quantum Eigensolver (VQE) | A hybrid quantum-classical algorithm used to compute ground and excited state energies on quantum hardware, reducing the computational cost of on-the-fly electronic structure calculations [2]. | Used in the SHARC-TEQUILA framework for nonadiabatic dynamics [2]. |
| Projected Quantum Momentum (PQM) | A correction term derived from the Exact Factorization framework that improves the description of electronic coherence in MQC dynamics [1]. | Part of a unified approach with the phase-correction for accurate nonadiabatic features [1]. |
| SHARC Software Package | A molecular dynamics program package designed for performing surface hopping dynamics, including nonadiabatic couplings [2]. | Integrated with the TEQUILA quantum computing framework for hybrid simulations [2]. |
| TEQUILA Framework | A quantum computing framework that allows for the construction and computation of generalized quantum algorithms, such as VQE [2]. | Used in conjunction with SHARC to offload electronic structure calculations to a quantum simulator or hardware [2]. |
Implementing MQC Dynamics: Hybrid Algorithms and Real-World Biomedical Applications
FAQs: Core Concepts and Setup
Q1: What is the specific value of VQE in mixed quantum-classical dynamics simulations for drug development? VQE is a hybrid algorithm that uses a quantum computer to prepare and measure trial quantum states representing molecular configurations, and a classical computer to optimize the parameters of those states [21]. Within mixed quantum-classical dynamics, its primary value lies in calculating key electronic properties on-the-fly during molecular dynamics simulations. This includes ground and excited state energies, energy gradients (forces), and nonadiabatic coupling vectors [22] [23]. These quantities are essential for accurately simulating nonadiabatic processes like photo-induced isomerization or electronic relaxation, which are critical for understanding photochemical reactions in drug development [22].
Q2: Which software frameworks are available for implementing mixed quantum-classical dynamics with VQE? Researchers can utilize integrated software frameworks that bridge molecular dynamics and quantum computing. A prominent example is the interface between the SHARC (Surface Hopping including Arbitrary Couplings) molecular dynamics package and the TEQUILA quantum computing framework [22] [23]. This integration allows for the use of VQE and related algorithms to compute electronic structure properties needed to propagate nuclear dynamics, enabling the study of photochemical processes in molecules like methanimine and ethylene [22].
Q3: What are the most significant bottlenecks when running VQE on current quantum hardware? The main bottlenecks are:
- Measurement Precision and Overhead: Achieving chemical precision (1.6 × 10⁻³ Hartree) requires a massive number of measurements ("shots") and is hampered by readout errors [24].
- Algorithmic Convergence: The classical optimizer in the VQE loop can get trapped in local minima or suffer from "barren plateaus" in the energy landscape, preventing convergence to the true ground state [21].
- Circuit Depth and Noise: The parameterized quantum circuits (ansätze) must be shallow enough to be executed on noisy hardware, which can limit their ability to accurately represent complex molecular states [21].
FAQs: Troubleshooting Common Experimental Problems
Q4: How can I improve the energy estimation accuracy of VQE on noisy devices? Techniques from advanced quantum measurement theory can significantly enhance accuracy. Research demonstrates that a combination of the following methods can reduce estimation errors by an order of magnitude [24]:
- Quantum Detector Tomography (QDT): Characterizes and mitigates readout errors, directly reducing the bias in energy estimates.
- Locally Biased Random Measurements: A type of "classical shadows" technique that reduces the number of shots required by prioritizing measurement settings that have a larger impact on the final energy estimation.
- Blended Scheduling: Executes circuits in a specific order to mitigate the impact of time-dependent noise on the hardware.
Q5: My VQE optimization is not converging. What are the potential causes and solutions? Non-convergence typically stems from the classical optimizer or the quantum hardware. The following table outlines common issues and corrective actions.
| Problem Area | Specific Issue | Corrective Action |
|---|---|---|
| Classical Optimizer | Poor choice of optimizer for the problem's noise landscape. | Switch to robust optimizers like SPSA or NFT, which are designed for noisy environments [25]. |
| Initial parameters are far from the solution. | Use classically computed parameters (e.g., from MP2 theory) as a starting point. | |
| Quantum Hardware & Circuit | Noise and errors overwhelming the energy signal. | Implement error mitigation techniques like Zero-Noise Extrapolation (ZNE) [26]. |
| The chosen ansatz is not expressive enough or is too deep. | Experiment with different ansätze (e.g., k-UpCCGSD [22]) and systematically reduce circuit depth where possible. |
Q6: What is the best strategy to manage the computational overhead of measuring complex molecular Hamiltonians? The high number of Pauli terms in molecular Hamiltonians is a major source of measurement overhead. The following strategies can help manage this cost [24]:
- Commuting Clique Grouping: Group Hamiltonian terms into sets of commuting Pauli operators that can be measured simultaneously. For example, one study reduced the number of measurement bases for an ethylene molecule from 73,089 to just 552 [22].
- Informationally Complete (IC) Measurements: Use measurement protocols that allow for the estimation of multiple observables from the same set of data, thereby increasing data efficiency [24].
Experimental Protocols and Data Presentation
Protocol: Implementing a VQE-Based Mixed Quantum-Classical Dynamics Simulation
This protocol outlines the key steps for simulating nonadiabatic molecular dynamics using VQE, based on the SHARC-TEQUILA integration [22].
- System Preparation: Define the molecular system and select an active space for the quantum chemical calculation.
- Ansatz Selection: Choose a parameterized quantum circuit (e.g., k-UpCCGSD [22]) for the VQE algorithm.
- Classical Initialization: Set initial nuclear coordinates and momenta (( \vec{R}0, \vec{P}0 )).
- VQE Execution Loop: For each classical timestep: a. Hamiltonian Construction: Build the electronic Hamiltonian in the qubit basis (e.g., via Jordan-Wigner transformation) using the current nuclear coordinates ( \vec{R}t ) [22]. b. State Preparation: Run VQE to find the parameters ( \vec{\theta}t ) that minimize the energy ( \langle \psi(\vec{\theta}t) | H(\vec{R}t) | \psi(\vec{\theta}t) \rangle ), preparing the ground (or excited) state wavefunction [22]. c. Observable Measurement: Measure the required electronic properties from the optimized state ( | \psi(\vec{\theta}t) \rangle ). This includes energies, forces (approximated via the Hellmann–Feynman theorem), and nonadiabatic coupling vectors [22].
- Nuclear Propagation: Feed the computed forces into the classical molecular dynamics propagator (e.g., Tully's surface hopping) to update the nuclear positions and momenta to ( \vec{R}{t+1}, \vec{P}{t+1} ) [22].
- Iteration: Repeat steps 4a-5 until the total simulation time is reached.
Diagram 1: VQE Molecular Dynamics Workflow
Performance Data: VQE vs. Other Quantum Algorithms
The following table summarizes a benchmarking study comparing the performance of VQE and quantum dynamics algorithms for solving a 1D advection-diffusion equation, providing a baseline for algorithmic performance on a 4-qubit system [25].
| Algorithm | Type | Key Principle | Reported Infidelity (Noiseless Simulator) | Reported Infidelity (Hardware) |
|---|---|---|---|---|
| VQE (Statevector) | Hybrid Variational | Maps timestep to a ground-state problem; classical parameter optimization [25]. | (\mathcal{O}(10^{-9})) [25] | Not tested in this study [25]. |
| Trotterization | Quantum Dynamics | Direct Hamiltonian simulation via decomposed time steps [25]. | (\mathcal{O}(10^{-5})) [25] | (\gtrsim \mathcal{O}(10^{-1})) [25] |
| VarQTE | Hybrid Variational | Variational simulation of imaginary time evolution [25]. | (\mathcal{O}(10^{-5})) [25] | (\gtrsim \mathcal{O}(10^{-1})) [25] |
| AVQDS | Hybrid Variational | Adaptive ansatz expansion for dynamics simulation [25]. | (\mathcal{O}(10^{-5})) [25] | (\gtrsim \mathcal{O}(10^{-1})) [25] |
The Scientist's Toolkit: Research Reagent Solutions
This table details essential software and methodological "reagents" for conducting mixed quantum-classical dynamics experiments with VQE.
| Item | Function in the Experiment | Technical Specification / Example |
|---|---|---|
| TEQUILA Framework | A quantum computing framework used to implement VQE and other variational algorithms for calculating electronic properties [22] [23]. | Integrated with SHARC; supports various ansätze and optimizers [22]. |
| SHARC Package | Molecular dynamics program that propagates nuclear trajectories, using electronic inputs from TEQUILA [22] [23]. | Implements Tully's fewest switches surface hopping and other methods [22]. |
| k-UpCCGSD Ansatz | A parameterized quantum circuit (ansatz) used to prepare trial wavefunctions in VQE. Offers a good compromise between accuracy and computational cost [22]. | A unitary coupled-cluster type ansatz; more efficient than UCCSD for some applications [22]. |
| Quantum Detector Tomography (QDT) | A calibration technique that characterizes readout errors on the quantum device, enabling the mitigation of bias in energy estimations [24]. | Implemented by measuring a set of pre-determined calibration circuits to build a noise model [24]. |
| Zero-Noise Extrapolation (ZNE) | An error mitigation technique that intentionally scales up circuit noise to extrapolate back to a zero-noise result [26]. | Demonstrated in code examples for VQE to improve energy estimation accuracy [26]. |
Diagram 2: Noise Mitigation Strategy Map
Troubleshooting Guides
Guide 1: Resolving Electronic Energy Calculation Failures
Problem: The Variational Quantum Eigensolver (VQE) fails to converge to the ground state energy during a dynamics trajectory.
Explanation: VQE convergence issues often stem from an inappropriate ansatz choice, noisy hardware, or an inadequate classical optimizer [2] [22]. This failure directly impacts force calculations and nuclear propagation.
Solution: Follow this systematic troubleshooting procedure:
- Verify Ansatz Initialization: Confirm the
k-UpCCGSDansatz is initialized with Hartree-Fock reference states from the classical computer. Check for orbital symmetry mismatches [22]. - Optimizer Adjustment: Switch from gradient-based optimizers to robust methods like
COBYLAorSPSAwhich are more noise-resilient on NISQ devices [2]. - Check Hamiltonian Transformation: Validate the Jordan-Wigner transformation of the molecular Hamiltonian. Ensure Pauli terms are grouped into commuting cliques to minimize quantum circuit evaluations [22].
Guide 2: Addressing Inaccurate Nonadiabatic Transitions
Problem: Surface hopping probabilities between electronic states are incorrect, leading to unphysical dynamics.
Explanation: This typically arises from inaccurate calculation of nonadiabatic coupling vectors (NACVs) or energy gaps between states [2] [1]. The penalty-based variational quantum deflation (VQD) method is sensitive to penalty weight selection.
Solution:
- Recalculate NACVs: Use the finite difference method to verify NACVs. On quantum hardware, ensure consistent measurement bases for off-diagonal elements of the energy and derivative operators [2].
- Adjust VQD Penalty Weight: Systematically increase the penalty weight in the VQD cost function to enforce orthogonality. However, avoid excessively high weights that cause numerical instabilities [2] [22].
- Validate State Orthogonality: For each new geometry, check the overlap
<ψ_i|ψ_j>between computed states. It should be below a defined threshold (e.g., 1x10⁻⁵) [22].
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of using the SHARC-TEQUILA pipeline over fully classical simulations?
A1: The pipeline leverages quantum computers to compute electronic properties that are computationally expensive for classical computers, such as ground and excited state energies. This is promising for achieving a "quantum advantage" in simulating photochemical dynamics for increasingly larger molecules, moving beyond the limitations of classical computational methods [2] [27].
Q2: Why are the Pulay force terms often neglected in the gradient calculations, and what are the implications?
A2: The full energy gradient includes the Hellmann-Feynman term and additional Pulay terms. In the current implementation, only the Hellmann-Feynman term is typically calculated for simplicity, as shown in the equation ∇ξE ≈ <Ψ(θ)| ∇ξĤ |Ψ(θ)> [22]. This approximation is common in initial quantum-classical dynamics studies but can introduce errors in the nuclear forces, especially when the wavefunction is not fully optimized with respect to the parameters θ. Users should be aware this may affect energy conservation in long-time dynamics [22].
Q3: Our simulations show poor electronic decoherence. How can this be improved within the SHARC-TEQUILA framework?
A3: Standard surface hopping methods like Tully's approach, which SHARC implements, are known to have inherent limitations in describing decoherence. Recent advances derived from the Exact Factorization framework, such as including a Projected Quantum Momentum (PQM) correction and a phase-correction term, have shown promise in more accurately capturing decoherence and phase evolution [1]. Investigating the integration of such corrections into the SHARC-TEQUILA feedback loop is an area of active research [1] [28].
Experimental Protocols & Data
Table 1: Key Electronic Properties and Computational Methods
| Electronic Property | Algorithm Used | Classical/Quantum Computation | Key Parameters |
|---|---|---|---|
| Ground State Energy | Variational Quantum Eigensolver (VQE) [2] [22] | Hybrid | Ansatz: k-UpCCGSD [22] |
| Excited State Energies | Variational Quantum Deflation (VQD) [2] [22] | Hybrid | Penalty weight, Orthogonality constraint |
| Energy Gradients (Forces) | Hellmann-Feynman Theorem [22] | Hybrid | Central difference (shift: 0.001 Å) |
| Nonadiabatic Couplings | Finite Difference / VQD outputs [2] | Hybrid | - |
| Transition Dipole Moments | VQE/VQD Wavefunctions [2] | Hybrid | - |
Table 2: Benchmark Systems for Validation
| Molecular System | Process Studied | Validation Criterion | Observed Performance |
|---|---|---|---|
| Methanimine | cis–trans photoisomerization [2] | Qualitative agreement with experiment & classical simulations | Correctly captured reaction pathways [2] |
| Ethylene | ultrafast electronic relaxation [2] | Reproduction of known relaxation dynamics | Qualitatively accurate dynamics observed [2] |
Protocol: Running a SHARC-TEQUILA Dynamics Simulation
Initialization:
- Classical Step: Generate initial nuclear coordinates and momenta. Perform a Hartree-Fock calculation to obtain one- and two-electron integrals for the current geometry
R(t)[22]. - Quantum Step: Transform the electronic Hamiltonian to a qubit operator using the Jordan-Wigner transformation. Prepare the
k-UpCCGSDansatz circuit on the quantum processor [22].
- Classical Step: Generate initial nuclear coordinates and momenta. Perform a Hartree-Fock calculation to obtain one- and two-electron integrals for the current geometry
Electronic Structure Calculation:
Property Evaluation:
Nuclear Propagation:
- The SHARC package propagates the classical nuclei for one timestep using the computed forces, typically via Tully's fewest switches surface hopping [2].
Loop: Repeat steps 1-4 for the desired number of dynamics steps, updating the Hamiltonian with the new geometry
R(t+Δt)at each step [2] [27].
Workflow Visualization
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Components
| Component | Function / Description | Role in the Pipeline |
|---|---|---|
| SHARC (Molecular Dynamics) | A software package for performing mixed quantum-classical nonadiabatic dynamics, implementing methods like surface hopping [2]. | Manages the entire dynamics workflow: propagates classical nuclei, decides hopping events, and calls the electronic structure solver. |
| TEQUILA (Quantum Computing) | A flexible quantum computing platform for constructing and minimizing generalized objective functions [2]. | Provides the interface to quantum hardware/simulators, runs VQE and VQD algorithms to solve the electronic structure problem. |
| VQE Algorithm | A hybrid algorithm to find the ground state of a system by varying parameters of a quantum circuit [2] [22]. | Computes the ground state energy and wavefunction at each nuclear geometry. |
| VQD Algorithm | An extension of VQE that uses penalty terms to compute excited states orthogonal to the ground state [2] [22]. | Computes excited state energies and wavefunctions needed for nonadiabatic transitions. |
| k-UpCCGSD Ansatz | A compact and efficient quantum circuit ansatz for wavefunction preparation [22]. | Defines the trial wavefunction form for VQE/VQD, balancing accuracy and computational cost. |
| Jordan-Wigner Transform | A method for mapping fermionic creation/annihilation operators to Pauli spin operators [22]. | Encodes the molecular electronic Hamiltonian into a form executable on a qubit-based quantum computer. |
This technical support center addresses the practical implementation challenges of mixed quantum-classical (MQC) dynamics in drug discovery research. As you work to simulate complex biological processes like protein-ligand binding and hydration, you will encounter unique obstacles at the intersection of quantum hardware, classical software, and algorithmic design. The following guides and FAQs are built upon recent experimental studies and are designed to help you diagnose and resolve specific issues, enabling more robust and reliable research outcomes.
Frequently Asked Questions & Troubleshooting Guides
Hardware & Algorithm Implementation
Q: My hybrid quantum-classical workflow is failing due to high quantum hardware noise. What mitigation strategies can I implement?
- Problem: Quantum processing units (QPUs) are prone to errors from decoherence and noise, which disrupts superposition and entanglement, leading to unreliable results in your protein-ligand simulations [29].
- Solution:
- Utilize Error Suppression Software: Integrate tools like Q-CTRL Fire Opal into your workflow on platforms such as Amazon Braket. These tools can actively suppress errors, leading to documented performance improvements in algorithms for applications like financial risk analysis and quantum network anomaly detection [30].
- Leverage Hybrid Jobs: Use the Amazon Braket Hybrid Jobs feature, which is designed to manage classical resources efficiently and handle job queuing for QPUs, reducing the impact of variable hardware conditions [30].
- Circuit Optimization: Prior to submission, analyze your quantum circuits for opportunities to reduce depth and gate count, thereby minimizing the window during which noise can affect the computation.
Q: How can I effectively scale my protein-ligand docking site identification algorithm to larger proteins?
- Problem: The available number of qubits is insufficient to represent all interaction sites for a large protein, limiting the scope of your research.
- Solution: The quantum algorithm for docking site identification is designed for scalability [31]. Its resource requirements are determined by the number of protein-ligand interaction sites, not the entire protein fold.
- Focus on Active Sites: Concentrate your quantum resources on modeling the key interaction sites within the protein's binding pocket.
- Modular Qubit Encoding: As detailed in the experimental protocol (see below), each interaction site (e.g., for hydrophobic or hydrogen bonding) is encoded with a dedicated qubit [31]. This means you can start with the most critical interactions and expand the model as more qubits become available.
- Future-Proofing: The algorithm architecture allows for the straightforward inclusion of additional interaction types (e.g., π-stacking, salt bridges) simply by adding more qubits to the quantum register per site [31].
Scientific Modeling & Validation
Q: My mixed quantum-classical dynamics simulation is producing inaccurate nonadiabatic transitions. What could be wrong?
- Problem: Traditional MQC methods like Ehrenfest dynamics or surface hopping often lack a rigorous mechanism for electronic decoherence, leading to incorrect transition probabilities between electronic states [1].
- Solution: Investigate and implement newly derived corrections from the Exact Factorization (XF) framework.
- Apply Advanced Corrections: Recent research has unified the treatment of decoherence and phase evolution by introducing two key corrections to the equations of motion: the Projected Quantum Momentum (PQM) and a previously unidentified phase-correction term [1].
- Implementation Check: Ensure your dynamics code incorporates these
ℏ-order terms, which have been validated on model systems like the double-arch geometry (DAG) and two-dimensional nonseparable (2DNS) models, showing improved accuracy in capturing nonadiabatic features such as Stückelberg oscillations [1].
Q: How can I validate that my quantum-computed protein hydration results are reliable?
- Problem: Results from a noisy quantum device may not reflect biological reality, making it risky to base drug discovery decisions on them.
- Solution: Employ a multi-layered validation strategy, as demonstrated in recent successful studies.
- Classical Cross-Verification: Always run parallel simulations using established classical methods (e.g., molecular dynamics with explicit water) on high-performance computing (HPC) clusters. Use this as a benchmark for your quantum results [32] [4].
- Experimental Validation: This is the gold standard. Follow the approach used by St. Jude and the University of Toronto: take the novel ligands generated or identified by your quantum-augmented pipeline and conduct in vitro binding assays. They successfully validated their quantum-derived molecules for the KRAS protein target, confirming real-world binding affinity [33].
- Quantum Simulator Baseline: Before running on a physical QPU, execute your algorithm on a noisy quantum simulator and a state-vector simulator (which provides an ideal, noise-free result) to establish an expected baseline and identify algorithm-specific versus hardware-induced errors [31].
Experimental Protocols & Workflows
Protocol 1: Quantum Algorithm for Protein-Ligand Docking Site Identification
This protocol details the methodology for identifying ligand docking sites on proteins using a modified Grover search algorithm [31].
1. Problem Encoding:
- Model: Represent the protein and ligand using an extended protein lattice model. Each amino acid in the protein chain comprises a set of finer-grained "interaction sites" [31].
- Qubit Encoding: Encode the chemical properties of each interaction site into qubits. For example, use two qubits per site to represent the presence or absence of:
- Hydrophobic interactions (
q_H) - Hydrogen bonding (
q_B)
- Hydrophobic interactions (
- State Preparation: The total quantum state for the protein (
|Protein>) is the tensor product of the quantum states of all its interaction sites. Similarly, the ligand's state (|Ligand>) is prepared as the tensor product of its interaction sites [31].
2. Algorithm Execution (Modified Grover Search):
- Step 1 - Create Superposition: Apply Hadamard gates to the qubits representing the protein's interaction sites, creating a uniform superposition to explore all possible binding regions simultaneously [31].
- Step 2 - Oracle Operation: The quantum oracle is designed to mark (flip the phase of) those states where the ligand's interaction pattern matches a segment of the protein's interaction pattern. This identifies potential docking sites [31].
- Step 3 - Amplification Operation: Apply amplitude amplification to increase the probability of measuring the correct docking site(s) identified by the oracle. The specific number of Grover iterations is determined by the search space size [31].
- Step 4 - Measurement: Measure the quantum state. The highest-probability outcomes correspond to the most likely docking sites for the ligand on the protein [31].
The workflow for this protocol is summarized in the diagram below:
Protocol 2: Hybrid Quantum-Classical Dynamics for Molecular Systems
This protocol outlines a general approach for MQC dynamics, where a quantum computer calculates electronic properties and a classical computer handles nuclear motion [2] [27].
1. Initialization:
- Classical System: Initialize the classical nuclear coordinates (
R₀) and momenta (P₀). - Quantum Subsystem: On the QPU, prepare the initial electronic wavefunction
|ψ₀>for the system at geometryR₀. This can be done using a Variational Quantum Eigensolver (VQE) to find the ground state [2] [27].
2. Time Propagation Loop:
- Step A - Quantum Force Calculation:
- Step B - Classical Nuclear Update:
- On the CPU, propagate the classical nuclear coordinates and momenta to time
t+Δtusing the forces obtained from the QPU. This can be done using a classical integrator like Velocity Verlet [27].
- On the CPU, propagate the classical nuclear coordinates and momenta to time
- Step C - Hamiltonian Update & Quantum State Propagation:
- Update the electronic Hamiltonian based on the new nuclear coordinates
R_{t+Δt}. - On the QPU, evolve the electronic wavefunction from
|ψ_t>to|ψ_{t+Δt}>. This can be achieved with a time-propagation algorithm like Time-Dependent Variational Quantum Propagation (TDVQP) [27].
- Update the electronic Hamiltonian based on the new nuclear coordinates
- Iterate: Repeat steps A-C until the desired simulation time is reached.
The following diagram illustrates this iterative feedback loop:
The table below lists key computational "reagents" and resources essential for building and running quantum simulations for drug discovery.
| Resource Name | Type / Function | Key Use-Case in Drug Discovery |
|---|---|---|
| Pasqal Quantum Processor [32] [4] | Neutral-atom quantum hardware (e.g., "Orion") | Executing quantum algorithms for precise placement of water molecules in protein pockets (hydration analysis) [32] [4]. |
| Variational Quantum Eigensolver (VQE) [2] | Hybrid quantum-classical algorithm | Calculating ground and excited state energies of molecular systems for dynamics simulations [2]. |
| Grover Search Algorithm [31] | Quantum search algorithm | Rapidly identifying potential protein-ligand docking sites within an unsorted interaction space [31]. |
| SHARC & TEQUILA [2] | Integrated software framework (SHARC for molecular dynamics, TEQUILA for quantum computing) | Propagating nonadiabatic molecular dynamics using electronic properties computed on a quantum computer [2]. |
| Amazon Braket [30] | Cloud-based quantum computing service | Managed access to multiple quantum hardware providers, simulators, and tools for building hybrid quantum-classical algorithms [30]. |
| Q-CTRL Fire Opal [30] | Error suppression/performance optimization software | Improving algorithm reliability and performance on noisy quantum hardware for tasks like financial modeling and cybersecurity [30]. |
Performance Data & Benchmarks
The table below summarizes key quantitative findings from recent research, providing benchmarks for your experiments.
| Study / System | Key Metric / Result | Method & Hardware Used |
|---|---|---|
| Protein Hydration (Pasqal & Qubit Pharmaceuticals) [32] [4] | First quantum algorithm used for a molecular biology task of this importance. Successfully implemented on the Orion neutral-atom quantum computer. | Hybrid quantum-classical pipeline for precise water molecule placement in protein pockets [32] [4]. |
| KRAS Ligand Discovery (St. Jude & University of Toronto) [33] | Quantum machine learning model outperformed purely classical models. Two real-world binding molecules identified and experimentally validated. | Combined classical and quantum machine learning model for ligand generation, followed by in vitro experimental validation [33]. |
| Mixed Quantum-Classical Dynamics (SHARC + TEQUILA) [2] | Achieved qualitatively accurate molecular dynamics for photoisomerization of methanimine and electronic relaxation of ethylene, aligning with experimental data. | Variational Quantum Eigensolver (VQE) and Variational Quantum Deflation (VQD) on a quantum computer to provide energies and couplings for classical surface hopping dynamics [2]. |
Frequently Asked Questions (FAQs)
1. What is the core challenge that active space embedding aims to solve? Active space embedding addresses the problem of strong electron correlation in molecular and solid-state systems. Accurate simulation of these systems requires methods that can capture complex electron entanglement, but such methods typically have exponentially scaling computational costs, limiting their application to small systems [34] [35]. Embedding overcomes this by dividing the problem, using accurate (and expensive) methods only on a small, chemically relevant "fragment" of the entire system [34].
2. How does quantum computing integrate with this embedding framework? In a hybrid quantum-classical embedding scheme, the classical computer handles the large environmental background of the system using efficient methods like Density Functional Theory (DFT). The quantum computer is then tasked with solving the embedded fragment Hamiltonian—a smaller, but strongly correlated quantum problem—for properties like ground and excited states using algorithms like the Variational Quantum Eigensolver (VQE) [34] [35] [2].
3. My hybrid dynamics simulation is failing. What are the most common sources of error? Errors in mixed quantum-classical dynamics often stem from inaccuracies in the electronic structure data fed into the classical nuclear propagation. Key quantities to scrutinize are [2]:
- Energy gradients (forces)
- Nonadiabatic coupling vectors between electronic states
- Transition dipole moments These are computed by the quantum end of the simulation (either classical electronic structure or a quantum computer), and inaccuracies here will lead to incorrect nuclear trajectories.
4. What practical applications exist for these methods in drug discovery? These techniques are being explored for computationally intense tasks in pharmaceutical research, including [4] [5]:
- Protein-ligand binding studies: Precisely modeling how drug candidates bind to their protein targets.
- Analysis of protein hydration: Determining the position and role of water molecules in protein pockets, which critically influences binding.
- Prediction of off-target effects: Identifying potential side effects early in the drug development process by simulating interactions with unintended biological targets.
Troubleshooting Guides
Issue 1: Inaccurate Spectral Predictions in Periodic Systems
Problem Description: When using periodic range-separated DFT embedding to predict optical spectra (e.g., for a defect in a solid like MgO), the absorption band positions show discrepancies compared to experimental results, even if emission peaks are accurate [34] [35].
Diagnosis and Solution: This is a known challenge where the method's performance is competitive but not perfect. The following table summarizes the diagnostic steps and solutions.
| Diagnostic Step | Possible Cause | Recommended Action |
|---|---|---|
| Analyze the fragment Hamiltonian. | The active space (selection of orbitals and electrons) is too small to capture the essential correlation effects. | Systematically increase the size of the active space and monitor the convergence of the excited-state energies [34] [35]. |
| Check the embedding potential. | The description of the interaction between the fragment and the periodic environment is incomplete. | Verify the implementation of the range-separated DFT protocol and the coupling between the quantum and classical codes [34]. |
| Benchmark against other methods. | Intrinsic error of the methodology for specific electronic configurations. | Compare results with state-of-the-art ab initio approaches to contextualize the level of accuracy [34]. |
Issue 2: Instability in Mixed Quantum-Classical Dynamics Propagation
Problem Description: Surface hopping molecular dynamics simulations (e.g., studying photo-isomerization) become unstable or produce unphysical results [2].
Diagnosis and Solution: Instabilities often trace back to the electronic properties calculated for the classical nuclear motion.
| Diagnostic Step | Possible Cause | Recommended Action |
|---|---|---|
| Inspect the quantum computed energies and forces. | Noisy or inaccurate evaluation of ground and excited state energies and gradients on the quantum computer. | For VQE-based workflows, ensure the underlying variational circuit is sufficiently optimized. Use error mitigation techniques to improve result quality [2]. |
| Validate the nonadiabatic couplings. | Incorrect calculation of the couplings between electronic states, which govern the "hops". | Confirm that the quantum algorithm (e.g., Variational Quantum Deflation for excited states) correctly computes these vectors. Cross-check with a small, classically solvable system [2]. |
| Review the classical MD parameters. | Incompatibility between the time step and the highly oscillatory quantum-derived data. | Reduce the molecular dynamics time step and check if the instability persists. |
Issue 3: Quantum Resource Limitations for the Fragment Solver
Problem Description: The quantum computation of the fragment's electronic structure exceeds the capabilities of current noisy hardware, for example, due to deep quantum circuits or long variational optimization times [34].
Diagnosis and Solution: This is a fundamental hardware limitation addressed with algorithmic and strategic choices.
| Diagnostic Step | Possible Cause | Recommended Action |
|---|---|---|
| Profile the quantum circuit. | The ansatz circuit for the fragment is too deep, leading to errors from qubit decoherence. | Investigate and employ more hardware-efficient or chemically inspired ansatzes that shallower circuits [34]. |
| Assess the active space size. | The fragment Hamiltonian is mapped to too many qubits, making the problem intractable. | Re-evaluate the chemical system to determine if a smaller, yet physically relevant, active space can be used without sacrificing key accuracy [35]. |
| Evaluate the hybrid algorithm. | The variational quantum algorithm requires too many circuit evaluations for convergence. | Leverage advanced classical optimizers designed for noisy environments and consider quantum algorithms with better convergence properties [34] [2]. |
Experimental Protocols & Methodologies
Protocol 1: Implementing Periodic rsDFT Embedding with a Quantum Solver
This protocol details the steps for accurately predicting the optical properties of a localized defect in a solid, such as the neutral oxygen vacancy in MgO [34] [35].
1. System Preparation:
- Model the Periodic Crystal: Set up a supercell model of the bulk material (e.g., MgO) containing the point defect.
- Choose a Basis Set: Select appropriate Gaussian-type orbital (GTO) basis sets for the atoms and a plane-wave cutoff for the DFT calculation within the GPW method [34].
2. Active Space Selection:
- Identify the Fragment: Locate the orbitals and electrons associated with the defect site. For the neutral oxygen vacancy in MgO, this involves the electrons left in the cavity by the removed oxygen atom.
- Define Active Orbitals: Select the fragment and a minimal number of surrounding orbitals that are chemically relevant to the electronic states of interest. This defines the active space [34].
3. Classical Embedding Calculation:
- Run Range-Separated DFT: Perform a periodic rsDFT calculation for the entire supercell. This provides the one-electron embedding potential, ( V_{uv}^{\text{emb}} ), which incorporates the interaction with the inactive environment [34] [35].
- Construct Fragment Hamiltonian: Build the embedded fragment Hamiltonian, ( \hat{H}^{\text{frag}} ), using the active orbitals. Its general form is [34]: [ \hat{H}^{\text{frag}} = \sum{uv} V{uv}^{\text{emb}} \hat{a}{u}^{\dagger} \hat{a}{v} + \frac{1}{2} \sum{uvxy} g{uvxy} \hat{a}{u}^{\dagger} \hat{a}{x}^{\dagger} \hat{a}{y} \hat{a}{v} ]
4. Quantum Computation:
- Map to Qubits: Transform the fermionic fragment Hamiltonian into a qubit Hamiltonian using a transformation such as Jordan-Wigner or Bravyi-Kitaev.
- Compute Electronic States: Use the VQE to compute the ground state energy. Employ the quantum Equation-of-Motion (qEOM) algorithm to compute the low-lying excited states and their transition properties [34].
- Extract Spectra: Use the computed energies and transition dipoles to predict the optical absorption and emission spectra.
The workflow for this protocol is visualized below.
Protocol 2: Setting Up Mixed Quantum-Classical Dynamics with SHARC and TEQUILA
This protocol outlines how to run nonadiabatic surface hopping dynamics using electronic structure data from a quantum computer [2].
1. Initialization:
- Define Molecular System: Specify the initial molecular geometry, nuclear velocities, and the electronic states of interest.
- Configure Quantum Solver: Select the variational quantum ansatz (parameterized quantum circuit) for the VQE and VQD algorithms.
2. Single-Point Electronic Structure Calculation:
- Compute Key Properties: For the current nuclear configuration, use the hybrid quantum-classical setup to calculate:
- Ground and excited state energies
- Energy gradients (for forces) for each relevant state
- Nonadiabatic coupling vectors between states
- Transition dipole moments
- Pass Data to MD Engine: Send these computed properties to the classical molecular dynamics program (SHARC).
3. Nuclear Propagation:
- Integrate Newton's Equations: Use the computed forces to propagate the classical nuclei by one time step.
- Determine Electronic State Hop: Based on the nonadiabatic couplings and the Tully's fewest switches surface hopping algorithm, decide if the trajectory hops to a different electronic state.
4. Loop and Analyze:
- Repeat steps 2 and 3 for the desired number of simulation steps.
- Analyze an ensemble of trajectories to obtain dynamical properties, such as reaction quantum yields or electronic relaxation lifetimes.
The logical flow of this simulation is described below.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key software and algorithmic "reagents" essential for implementing the active space embedding and mixed quantum-classical dynamics methods discussed.
| Item Name | Type | Primary Function |
|---|---|---|
| CP2K [34] [35] | Software Package | A powerful classical molecular dynamics package, used here to perform the periodic rsDFT calculation and generate the embedding potential for the environment. |
| Qiskit Nature [34] [35] | Software Library | An extension of the Qiskit quantum computing SDK, used to map the fragment Hamiltonian to a qubit representation and run quantum algorithms like VQE and qEOM. |
| Variational Quantum Eigensolver (VQE) [34] [2] | Quantum Algorithm | A hybrid algorithm used to find the ground-state energy of the fragment Hamiltonian by optimizing a parameterized quantum circuit on a quantum processor with the help of a classical optimizer. |
| Quantum Equation-of-Motion (qEOM) [34] | Quantum Algorithm | An extension of VQE used to compute the excited-state spectrum of the fragment Hamiltonian, which is crucial for predicting optical properties. |
| SHARC [2] | Software Package | A molecular dynamics program designed for nonadiabatic dynamics simulations, handling the propagation of nuclei using Tully's surface hopping and classical forces. |
| TEQUILA [2] | Software Framework | A quantum computing framework that facilitates the construction and execution of variational quantum algorithms, integrating with SHARC to provide the necessary electronic structure data. |
FAQs: Mixed Quantum-Classical Dynamics in Pharmaceutical Workflows
Q1: How can Mixed Quantum-Classical (MQC) dynamics methods address the computational cost of simulating quantum effects in drug-sized molecules?
MQC methods tackle this by partitioning the system: the electronic subsystem (where quantum effects are critical) is treated quantum mechanically, while the nuclear dynamics are treated classically. This integration captures essential nonadiabatic effects like electronic transitions during chemical reactions, but the quantum chemistry calculations remain computationally expensive. To mitigate this, hybrid quantum-classical algorithms can be implemented where the required electronic properties (ground and excited state energies, gradients, nonadiabatic coupling vectors) are computed using quantum computing hardware, specifically the Variational Quantum Eigensolver (VQE), while classical computers handle the nuclear trajectory propagation [2] [27].
Q2: What are the fundamental shortcomings of traditional MQC methods like Ehrenfest dynamics and surface hopping, and how are modern approaches overcoming them?
Traditional methods suffer from two key shortcomings: the absence of a reliable mechanism for electronic decoherence and the lack of a rigorous treatment of phase evolution between adiabatic states. Modern approaches derived from the Exact Factorization (XF) formalism are providing more rigorous solutions. The XF framework reveals that a projected quantum momentum (PQM) correction is essential for describing electronic coherence, and it has also identified a previously overlooked phase-correction term. The combined inclusion of both PQM and phase corrections within the equations of motion provides a unified and balanced description of both decoherence and phase dynamics without relying on semiclassical approximations [1].
Q3: In the context of covalent inhibitor design, how can new experimental data analysis methods accelerate the identification of selective compounds?
The design of safe covalent inhibitors requires optimizing the balance between binding affinity and reactivity to minimize off-target effects. A new proteome-wide method called COOKIE-Pro (Covalent Occupancy Kinetic Enrichment via Proteomics) addresses this by simultaneously measuring the binding strength (affinity) and reaction speed (inactivation rate) of a covalent drug candidate against thousands of proteins in a single experiment. This comprehensive, unbiased map of drug-protein interactions allows medicinal chemists to prioritize compounds that are potent due to specific target binding, rather than inherent, non-specific reactivity, thereby accelerating the design of safer therapeutics [36].
Q4: What is the practical role of hybrid quantum-classical computing pipelines in current drug discovery for problems like prodrug activation?
Hybrid quantum-classical pipelines are being tailored for real-world drug design challenges, moving beyond proof-of-concept studies. A practical application involves using the Variational Quantum Eigensolver (VQE) to compute the Gibbs free energy profile for prodrug activation, a process involving covalent bond cleavage. In one case study, the pipeline accurately simulated the carbon-carbon bond cleavage for a β-lapachone prodrug, calculating the reaction energy barrier—a critical factor in determining if activation occurs under physiological conditions. The workflow integrated a solvent model (ddCOSMO) to simulate the aqueous environment of the human body, demonstrating the potential of quantum computing to handle essential steps in real-world drug design tasks [37].
Troubleshooting Common Implementation Challenges
Troubleshooting Prodrug Activation Workflows
Table 1: Common Issues in Prodrug Activation Simulations
| Challenge | Potential Cause | Solution |
|---|---|---|
| Inaccurate Gibbs free energy barrier for bond cleavage [37]. | Inadequate treatment of solvent effects or active space selection in quantum computation. | Implement a polarizable continuum model (PCM) like ddCOSMO to simulate solvation. Use active space approximation (e.g., 2 electrons in 2 orbitals) for the reaction center to make quantum computation feasible on current hardware. |
| Quantum device noise leads to unreliable energy calculations [37]. | Intrinsic noise and limited coherence times in NISQ-era quantum processors. | Apply readout error mitigation techniques. Use a hardware-efficient ansatz with a minimal number of layers in the VQE algorithm to reduce circuit depth and susceptibility to noise. |
| Computational cost of simulating full reaction pathway is prohibitive. | The exponential scaling of fully quantum mechanical methods. | Adopt a hybrid MQC dynamics approach. Use quantum computing only for the electronic structure calculations of key states (reactants, transition states, products) along the reaction path, while relying on classical methods for conformational sampling and dynamics [2]. |
Troubleshooting Covalent Inhibitor Design
Table 2: Common Issues in Covalent Inhibitor Design and Validation
| Challenge | Potential Cause | Solution |
|---|---|---|
| Off-target toxicity of covalent inhibitors [36]. | Lack of proteome-wide understanding of the inhibitor's binding affinity and reactivity. | Utilize COOKIE-Pro profiling to measure occupancy and inactivation rates across thousands of proteins, identifying off-targets early. Use this data to guide structural optimization for selectivity. |
| Difficulty accurately simulating covalent bond formation dynamics [37]. | The quantum mechanical nature of bond formation and breaking is challenging for purely classical force fields. | Employ a hybrid QM/MM (Quantum Mechanics/Molecular Mechanics) simulation setup. The covalent binding site is treated quantum mechanically (potentially using a quantum computer via VQE), while the rest of the protein and solvent is treated with molecular mechanics. |
| Low lead compound potency during optimization [38]. | Insufficient interaction with the target protein's active site. | Implement structure-based drug design (SBDD). Start from a known, moderate-affinity inhibitor (e.g., via drug repurposing screens) and perform rational, iterative cycles of structural optimization guided by the target's crystal structure. |
Experimental Protocols
Protocol: Structure-Based Design of a Covalent hCES2A Inhibitor
This protocol is based on the discovery of orally active serine-targeting covalent inhibitors for ameliorating irinotecan-triggered gut toxicity [38].
1. Initial Hit Identification:
- Perform a repurposing screen of FDA-approved drugs (e.g., 2000 compounds) against the target enzyme, human carboxylesterase 2A (hCES2A).
- Identify a moderate, reversible inhibitor as a starting point for optimization (e.g., Donepezil).
2. Structure-Based Optimization (Two Rounds):
- Use the crystal structure of the target enzyme (hCES2A) to guide the design of derivatives with improved binding affinity.
- Synthesize and assay these derivatives to identify a lead reversible inhibitor (e.g., compound B7).
3. Design of Covalent Inhibitors:
- Rationally design a series of covalent derivatives (e.g., carbamate compounds) based on the lead reversible inhibitor structure. These derivatives should feature an electrophilic "warhead" capable of covalently modifying the catalytic serine residue of the target.
- Synthesize and biologically assay the covalent compounds.
4. In Vitro and In Vivo Validation:
- Determine the half-maximal inhibitory concentration (IC₅₀) and specificity of the top covalent inhibitor (e.g., compound C3).
- Assess the compound's selectivity via proteome-wide profiling (e.g., using a method like COOKIE-Pro) [36].
- Evaluate the inhibitor's safety profile, metabolic stability, and intestinal exposure.
- Finally, validate its efficacy for ameliorating drug-triggered toxicity in relevant preclinical models, such as human intestinal organoids and tumor-bearing mice [38].
Protocol: Hybrid Quantum-Classical Pipeline for Prodrug Activation
This protocol outlines the use of a hybrid quantum-classical computing pipeline to calculate the Gibbs free energy profile of a prodrug activation reaction [37].
1. System Preparation and Conformational Optimization:
- Select the molecular system, including the key states along the reaction coordinate for covalent bond cleavage (e.g., reactant, transition state, product).
- Perform a conformational optimization of these structures using classical computational methods to obtain their minimum energy geometries.
2. Active Space Selection:
- Define the active space for the quantum computation. For feasibility on current hardware, this typically involves selecting the most relevant valence electrons and orbitals involved in the bond-breaking/forming process (e.g., a 2-electron/2-orbital active space).
3. Solvation Model Setup:
- Configure a solvation model to simulate the physiological environment. For example, use the ddCOSMO model within the polarizable continuum model (PCM) framework.
4. Hybrid Quantum-Classical Energy Calculation:
- For each key molecular structure (from Step 1), compute the single-point energy using a hybrid approach:
- Quantum Subroutine: Use the Variational Quantum Eigensolver (VQE) on a quantum processor (or simulator) to solve for the electronic energy within the selected active space. Employ a hardware-efficient ansatz and readout error mitigation.
- Classical Subroutine: Use classical methods (e.g., Hartree-Fock) to compute the thermal Gibbs corrections (entropy, enthalpy) for the free energy profile.
5. Energy Profile Construction and Analysis:
- Combine the quantum-computed electronic energies with the classically computed thermal corrections and solvation energies to construct the final Gibbs free energy profile for the reaction.
- Analyze the energy barrier to determine the feasibility of the reaction under physiological conditions.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagents and Computational Tools
| Item/Tool Name | Function/Application | Specification Notes |
|---|---|---|
| COOKIE-Pro Profiling [36] | Proteome-wide identification of off-target interactions and kinetic parameters (affinity & reactivity) for covalent inhibitors. | Requires a mass spectrometer for analysis. The "chaser" probe is a critical reagent. |
| hCES2A Enzyme Assay [38] | In vitro evaluation of inhibitor potency and specificity against the human carboxylesterase 2A target. | Used to determine IC₅₀ values. Can utilize recombinantly expressed enzyme. |
| Human Intestinal Organoids [38] | Preclinical model for validating the efficacy of compounds in ameliorating drug-induced gut toxicity. | Provides a more human-relevant model than simple cell lines for assessing biological effects. |
| Variational Quantum Eigensolver (VQE) [2] [37] | Hybrid quantum-classical algorithm for computing molecular ground state energies on noisy quantum hardware. | Typically implemented with a hardware-efficient ansatz and error mitigation for NISQ devices. |
| Polarizable Continuum Model (PCM) [37] | Implicit solvent model for simulating solvation effects in quantum chemical calculations of molecules in solution. | The ddCOSMO model is a specific type of PCM used for accuracy in energy calculations. |
| SHARC Molecular Dynamics [2] | A program package for performing nonadiabatic molecular dynamics simulations, such as surface hopping. | Can be integrated with quantum computing frameworks (e.g., TEQUILA) for the electronic structure part. |
| TEQUILA Framework [2] | A quantum computing platform for developing and executing hybrid quantum-classical algorithms. | Used in conjunction with classical MD packages like SHARC for mixed dynamics simulations. |
Solving Key Challenges: Decoherence, Force Accuracy, and Algorithmic Error Management
Frequently Asked Questions (FAQs)
1. What are the core shortcomings that PQM and phase-correction terms address in mixed quantum-classical dynamics? Mixed quantum-classical methods often struggle with two fundamental issues: the absence of a reliable mechanism for electronic decoherence and the lack of a rigorous treatment of phase evolution between adiabatic states. The PQM correction directly addresses electronic coherence, while the newly identified phase-correction term governs the proper phase evolution of the Born–Oppenheimer coefficients. Their combined inclusion provides a balanced description of both decoherence and phase dynamics without relying on semiclassical approximations [1].
2. From which theoretical framework do these correction terms originate? Both the PQM and phase-correction terms are derived rigorously from the Exact Factorization (XF) formalism. The derivation reveals that the phase-correction appears alongside the PQM correction within the order of ℏ, offering a unified account of their effects on the nuclear force [1].
3. In the equation of motion, what is the mathematical relationship between the Ehrenfest term, the PQM term, and the phase term?
The total time evolution of the BO coefficients is given by a sum of distinct contributions:
C˙j = C˙j^Eh + C˙j^QM + C˙j^PQM + C˙j^Div + C˙j^Ph
Here, C˙j^Eh is the standard Ehrenfest term, C˙j^PQM is the Projected Quantum Momentum term, and C˙j^Ph is the newly identified phase-correction term [1].
4. What quantitative improvements can I expect from implementing these corrections? Benchmark tests on model systems demonstrate that the new formulations capture key nonadiabatic features with high accuracy. This includes the reproduction of characteristic patterns such as Stückelberg oscillations, correct intermediate-time electronic coherence, and accurate nuclear-density distributions [1].
Troubleshooting Guides
Issue 1: Unphysical Results in Electronic Population Transfer
Problem: Your simulations show population transfers or coherence patterns that deviate from exact quantum results, particularly in the intermediate to long-time dynamics.
Solution:
- Verify Term Inclusion: Ensure both the PQM term (
C˙j^PQM) and the phase-correction term (C˙j^Ph) are included in your electronic equation of motion. Using only one may result in an unbalanced and inaccurate description [1]. - Check Coupling to Nuclei: The corrections involve terms that couple electronic evolution to the nuclear wavefunction via the total nuclear quantum momentum (
𝓟ν). Confirm that this quantum momentum is correctly calculated and projected. The PQM term specifically involves a projection onto the electronic subspace [1]. - Benchmark on Simple Models: Validate your implementation on standard model systems like the double-arch geometry (DAG) or two-dimensional nonseparable (2DNS) models, where the correct nonadiabatic features are well-known [1].
Issue 2: Instability in Nuclear Force Calculation
Problem: The nuclear trajectories become unstable when the new corrections are added to the force expression.
Solution:
- Inspect the Force Expression: The exact classical nuclear force in the XF framework is
𝐅ν = -∇νϵ~ + A˙ν. The corrections influence this force through the time-dependent vector potentialAνand the scalar potentialϵ~, which now incorporate effects from the ENC operatorsH^ENC(1)andH^ENC(2)[1]. - Review Numerical Differentiation: The term
A˙νrepresents a total time derivative. Ensure that its numerical integration is stable. Consider using robust numerical differentiation schemes with appropriate time steps. - Monitor the Quantum Potential: In related hydrodynamic formulations, neglecting the quantum potential (
V_Q) and higher-order gradients is a common step to derive mixed quantum-classical models. While this simplifies the model, be aware that it is an approximation. Instabilities may arise if the system dynamics are sensitive to these neglected quantum effects [28].
Issue 3: Implementing Corrections on Hybrid Quantum-Classical Hardware
Problem: Errors propagate and compound when running mixed quantum-classical dynamics on a quantum computer, where the electronic subsystem is offloaded to quantum hardware.
Solution:
- Error Budget Analysis: The algorithm inherits inaccuracies from its constituent parts: circuit compression, time evolution approximation, and the classical propagator. Model coherent error on the wavefunction as
|ψ̃〉 = √(1-I²)|ψ〉 + I|φ〉, whereIis the infidelity, and track how this error propagates into measured observables and subsequent classical steps [27]. - Mitigate Observable Measurement Error: Inaccuracies in measuring observables on the quantum computer (like the energy gradient used for the classical force) directly impact the quality of the classical propagation. Employ error mitigation techniques to improve the accuracy of these measurements [27].
- Use a Modular Algorithm: Implement a modular algorithm structure where the choice of ansatz, time evolution method, and classical integrator are replaceable. This allows you to test different components for their robustness to noise and error [27].
Experimental Protocols & Data
Protocol: Benchmarking PQM and Phase-Correction on a Model System
This protocol outlines how to validate an implementation of the PQM and phase-correction terms.
1. Objective: To verify that the new mixed quantum-classical equations of motion accurately reproduce key nonadiabatic features.
2. Materials (Computational Models):
- System: A one- or two-dimensional model system with a nonadiabatic coupling, such as the double-arch geometry (DAG) or two-dimensional nonseparable (2DNS) model [1].
- Initial State: A well-defined nuclear wavepacket on a specific adiabatic surface.
- Reference Data: Exact quantum results for the system, if available.
3. Methodology:
- Propagation: Propagate the coupled electronic-nuclear system using the new equations of motion derived from the Exact Factorization.
- Term Comparison: Run comparative simulations:
- Simulation A: With only the Ehrenfest term (
C˙j^Eh). - Simulation B: With Ehrenfest and PQM terms.
- Simulation C: With Ehrenfest, PQM, and phase-correction terms.
- Simulation A: With only the Ehrenfest term (
- Observables: Track the following key observables over time:
- Electronic population dynamics.
- Nuclear density distribution at the final simulation time.
4. Data Analysis: Compare the results of simulations A, B, and C against exact quantum benchmarks. The successful implementation will show that Simulation C most accurately captures effects like Stückelberg oscillations and correct final nuclear densities [1].
Quantitative Data from Benchmark Studies
Table 1: Key Correction Terms in the Electronic Equation of Motion [1]
| Term Name | Mathematical Expression (Simplified) | Primary Physical Effect |
|---|---|---|
Ehrenfest (C˙j^Eh) |
- (i/ℏ)ε_j C_j - Σ (P_ν/M_ν) · d_ν,jk C_k |
Mean-field evolution, lacks decoherence. |
PQM (C˙j^PQM) |
- Σ (𝓟_ν / M_ν) · (Re[ D_ν,jk ]) C_k |
Describes electronic coherence. |
Phase (C˙j^Ph) |
- Σ (𝓟_ν / M_ν) · (Im[ D_ν,jk ]) C_k |
Governs proper phase evolution. |
Table 2: Expected Performance Outcomes from Correction Implementation [1]
| Feature | Without PQM/Phase Corrections | With PQM & Phase Corrections |
|---|---|---|
| Stückelberg Oscillations | Not captured or inaccurate | Accurately reproduced |
| Intermediate-time Coherence | Over- or under-estimated | Correctly described |
| Nuclear Density Distribution | Deviates from benchmark | High accuracy agreement |
Research Reagent Solutions
Table 3: Essential Computational Tools for Method Implementation
| Tool / Resource | Function in Research | Relevant Context |
|---|---|---|
| Exact Factorization (XF) Formalism | Provides the rigorous theoretical foundation for deriving the PQM and phase-correction terms. | Core framework for deriving new mixed quantum-classical equations [1]. |
| Model Systems (e.g., DAG, 2DNS) | Serve as standardized benchmarks for validating the accuracy of the dynamics. | Used to demonstrate the method captures key nonadiabatic features [1]. |
| Variational Quantum Algorithms (VQAs) | Used on quantum hardware to solve for the electronic state in a hybrid quantum-classical setup. | Enables mixed quantum-classical dynamics on near-term quantum computers [27] [2]. |
| Polarizable Continuum Model (PCM) | Models solvation effects for chemical reactions in drug design applications. | Critical for realistic Gibbs free energy calculations in prodrug activation studies [39]. |
| Quantum Machine Learning (QML) | Leverages quantum computing to process high-dimensional data more efficiently. | Used in life sciences for tasks like predicting patient response or analyzing liquid biopsies [5]. |
Workflow and Conceptual Diagrams
Diagram: Mixed Quantum-Classical Dynamics with Advanced Corrections
Title: MQC Dynamics with PQM & Phase Correction
Diagram: PQM & Phase Correction in Electronic Evolution
Title: Electronic State Evolution with Corrections
Mitigating Algorithmic Errors in Variational Quantum Time Propagation
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary sources of algorithmic error in variational quantum time propagation? The main sources include ill-conditioned equations of motion in overparameterized ansätze, which lead to numerical instabilities, and the exponential concentration of cost function gradients (barren plateaus) due to noise, which severely limits the trainability of parameters [40] [41]. Furthermore, in mixed quantum-classical dynamics, errors can propagate between the quantum and classical subsystems through inaccurate observable measurements, affecting the entire simulation's fidelity [27].
FAQ 2: Can standard Error Mitigation (EM) techniques resolve the issue of cost function concentration? For a broad class of EM strategies—including Zero Noise Extrapolation, Virtual Distillation, and Probabilistic Error Cancellation—the exponential cost concentration typically cannot be resolved without committing exponential resources elsewhere. Some protocols, like Virtual Distillation, can even make it harder to resolve cost function values. However, protocols like Clifford Data Regression (CDR) have shown potential to aid training in settings where cost concentration is not too severe [41].
FAQ 3: How can numerical stability be improved for overparameterized wavefunction ansätze? An effective method is the adaptive time-dependent Variational Monte Carlo (atVMC) approach. It uses the local-in-time error (LITE) to dynamically identify and evolve only the most relevant variational parameters at each time step. This selective evolution reduces the dimensionality of the parameter space, mitigates ill-conditioning, and lessens the need for strong, bias-inducing regularization [40].
FAQ 4: What role does the "quantum momentum" play in mixed quantum-classical dynamics?
Within the Exact Factorization (XF) framework, the projected quantum momentum (PQM) and a related phase-correction term are crucial for a unified and rigorous description of electronic decoherence and phase evolution. These \(\hbar\)-order terms provide a more accurate nuclear force and improve the description of electronic coherence in nonadiabatic processes [1].
FAQ 5: Are there classical machine learning methods that can help with variational quantum optimization? Yes, methods like the Variational Generative Optimization Network (VGON) can learn to map simple random inputs to high-quality solutions for quantum problems. This approach has been shown to help in avoiding barren plateaus and in finding ground states of many-body quantum systems without the trainability issues that often hamper standard hybrid quantum-classical algorithms [42].
Troubleshooting Guides
Issue: Ill-Conditioned Equations of Motion in tVMC
- Problem Description: When using highly expressive ansätze (e.g., neural-network quantum states) in tVMC simulations, the quantum Fisher matrix (or overlap matrix) can become ill-conditioned. This manifests as exploding parameter updates or a complete failure of the ODE solver.
- Diagnosis Steps:
- Monitor the condition number of the quantum Fisher matrix
\(S_{k,k'}\)during simulation. A sharply rising condition number indicates ill-conditioning [40]. - Check if the problem persists with stronger regularization. If it does, the ansatz is likely overparameterized for the specific dynamics.
- Monitor the condition number of the quantum Fisher matrix
- Solution Protocols:
- Protocol 1 (Adaptive Parameter Selection): Implement the atVMC algorithm [40].
- Procedure:
- At each time step
\(t\), compute the LITE and the contribution of each parameter to its reduction. - Rank parameters by their "relevance."
- Evolve only a subset of the most relevant parameters such that the LITE remains below a predefined threshold
\(\eta\). - Re-evaluate the active parameter set at the next time step.
- At each time step
- Procedure:
- Protocol 2 (Regularization and Benchmarking):
- Use a Tikhonov regularization when solving the linear system
\(i\hbar S \dot{\alpha} = F\)[40]. - Benchmark the adaptive method against the fully-parameterized simulation on small, tractable systems to verify that accuracy is maintained while stability is improved.
- Use a Tikhonov regularization when solving the linear system
- Protocol 1 (Adaptive Parameter Selection): Implement the atVMC algorithm [40].
Issue: Barren Plateaus and Poor Trainability
- Problem Description: The cost function landscape becomes flat, leading to exponentially vanishing gradients with respect to the parameters as the system size increases. This makes optimization infeasible.
- Diagnosis Steps:
- Plot the variance of the cost function gradient over multiple random parameter initializations. An exponential decay with system size confirms a barren plateau.
- Determine if the problem is inherent to the ansatz or exacerbated by hardware noise.
- Solution Protocols:
- Protocol 1 (Careful Application of Error Mitigation):
- Protocol 2 (Classical Generative Modeling for Initialization):
- Use the VGON model to find good initial parameters on classical hardware [42].
- Procedure:
- Train the VGON (a variational autoencoder-based model) to output parameters that minimize the target cost function (e.g., energy).
- Sample from the trained decoder to obtain high-quality initial parameter sets for the quantum circuit, potentially bypassing the barren plateau region encountered by random initialization.
Issue: Error Propagation in Mixed Quantum-Classical Loops
- Problem Description: In algorithms like TDVQP, where a classical nuclear trajectory and a quantum electronic state are co-evolved, errors in quantum measurements (due to finite sampling or hardware noise) feed back into the classical force calculation, leading to drifts in the dynamics [27].
- Diagnosis Steps:
- Run the simulation with increasing numbers of measurement shots. If the results converge, the error is likely due to shot noise.
- Compare the simulated dynamics for a simple, benchmarked model (like the Shin-Metiu model) against known accurate results to identify deviations [27].
- Solution Protocols:
- Protocol 1 (Robust Integrator and Observables):
- Use a stable classical integrator (e.g., Velocity Verlet) for the nuclear motion [27].
- Where possible, use observables that are less sensitive to specific quantum state errors.
- Protocol 2 (Error-Aware Propagation):
- Procedure:
- Characterize the measurement error for your key observables (e.g., the energy gradient/force).
- Model the error propagation through the classical integrator. This can help in understanding the uncertainty in the nuclear trajectory.
- If using a p-VQD-style step for quantum state propagation, ensure the infidelity of the compressed state is sufficiently low to prevent significant error accumulation [27].
- Procedure:
- Protocol 1 (Robust Integrator and Observables):
Table 1: Error Mitigation Technique Profiles for Variational Algorithms. PECC = Probabilistic Error Cancellation, ZNE = Zero Noise Extrapolation, CDR = Clifford Data Regression, AFT = Algorithmic Fault Tolerance.
| Technique | Reported Error Reduction / Improvement | Key Limitation / Cost | Suitability for Time Propagation |
|---|---|---|---|
| Adaptive tVMC (atVMC) [40] | Significantly improves numerical stability; enables reliable simulation with expressive ansätze. | Requires LITE calculation and parameter ranking. | High; specifically designed for time evolution. |
| Clifford Data Regression (CDR) [41] | Can aid training where cost concentration is not severe. | Limited efficacy for deep circuits/severe noise; requires classical simulation for training. | Medium; potential for parameter training. |
| Virtual Distillation [41] | Can improve state purity for expectation values. | Can worsen the resolvability of cost function values for training. | Low; may hinder trainability. |
| ZNE & PECC [41] | Effective for mitigating expectation value errors. | Cannot resolve exponential cost concentration without exponential resource overhead. | Medium; for observable measurement, not for training. |
| Algorithmic Fault Tolerance (AFT) [43] | Reduces QEC overhead by 10-100x in simulations. | Requires specific hardware flexibility (e.g., neutral-atom systems). | High (long-term); for fault-tolerant execution. |
Table 2: Representative Error Rates and Algorithmic Performance in Quantum Simulations.
| System / Method | Metric | Reported Value / Performance | Context / Implication |
|---|---|---|---|
| State-of-the-Art Hardware | Error per operation [44] | ~0.000015% | A lower bound for algorithmic error to be meaningful. |
| Google Willow Chip | Logical calculation time [44] | ~5 minutes for a benchmark | Suggests time propagation circuits must be short-depth. |
| TDVQP Algorithm | Fidelity in short-time evolution [27] | Performs well | Qualitative results retained for longer times, but errors accumulate. |
| VGON for Optimization | Barren Plateaus [42] | Avoided for 18-spin model ground state | Classical generative models can provide a promising initialization. |
Experimental Protocols
Protocol: Benchmarking with the Transverse-Field Ising Model (1D)
This protocol is adapted from the benchmarking procedure in the atVMC study [40].
- Objective: To test the stability and accuracy of a variational quantum time propagation algorithm against a well-known model, using an overparameterized ansatz.
- System Hamiltonian:
\[ \hat{H} = -J \sum_{\langle i, j\rangle} \hat{\sigma}^z_i \hat{\sigma}^z_j - h \sum_i \hat{\sigma}^x_i \] - Initialization: Prepare the system in the ground state of the Hamiltonian with a specific field strength
\(h_i\). - Dynamics: Perform a quantum quench by suddenly changing the field strength to
\(h_f\)and propagating the state in time. - Variational Ansatz: Use a expressive ansatz such as a spin-Jastrow wave function or a Restricted Boltzmann Machine (RBM) neural-network quantum state.
- Methods Comparison:
- Standard tVMC: Evolve all parameters with standard regularization.
- Adaptive tVMC (atVMC): Use the LITE to dynamically select a subset of parameters for evolution.
- Key Metrics:
- Stability: Monitor the condition number of the equations of motion.
- Accuracy: Track observables like magnetization
\(\langle \sigma^z \rangle\)and compare with exact diagonalization or Density Matrix Renormalization Group (DMRG) results. - Efficiency: Record the number of parameters actively evolved per time step in atVMC.
Protocol: Nonadiabatic Molecular Dynamics with TDVQP
This protocol is based on the implementation for the modified Shin-Metiu model [27].
- Objective: To simulate the nonadiabatic dynamics of a molecule by coupling a quantum electronic subsystem (on a quantum computer) to a classical nuclear trajectory.
- System Setup: The Shin-Metiu model, a standard benchmark for nonadiabatic dynamics, partitioned into a classical nucleus and a quantum electron treated in a local diabatic basis.
- Algorithm Workflow:
- Initialization: At
\(t=0\), prepare the electronic wavefunction\(|\psi_0\rangle\)on the quantum processor corresponding to the initial nuclear coordinates\(\vec{q}_0\). - Observable Measurement: Measure the required observables
\(\{\hat{O}^{(s)}_0\}\)(e.g., forces) from\(|\psi_0\rangle\). - Classical Propagation: Update the nuclear coordinates to
\(\vec{q}_1\)using an integrator (e.g., Velocity Verlet) and the measured forces. - Quantum Propagation: Evolve the electronic state from
\(|\psi_0\rangle\)to\(|\psi_1\rangle\)using a time step under the Hamiltonian\(\hat{H}(\vec{q}_0)\). On hardware, this is done via a compressed circuit found by a p-VQD step. - Iteration: Repeat steps 2-4, using the new Hamiltonian
\(\hat{H}(\vec{q}_1)\)for the next quantum propagation step.
- Initialization: At
- Error Analysis:
- Quantify the coherent error in the wavefunction as infidelity
\(I\). - Track how this error propagates into the observable measurements and subsequently affects the classical nuclear trajectory [27].
- Quantify the coherent error in the wavefunction as infidelity
Workflow and Schematic Diagrams
Research Reagent Solutions
Table 3: Essential Software and Algorithmic "Reagents" for Variational Quantum Dynamics Research.
| Item Name | Type | Primary Function | Key Reference / Implementation |
|---|---|---|---|
| Local-in-Time Error (LITE) | Diagnostic Metric | Quantifies the local deviation between variational and exact evolution; core of adaptive schemes. | [40] |
| Adaptive tVMC (atVMC) | Algorithm | Dynamically controls ansatz expressivity during evolution to maintain stability and accuracy. | [40] |
| Time-Dependent VQP (TDVQP) | Algorithm | A modular meta-algorithm for mixed quantum-classical dynamics on near-term quantum computers. | [27] |
| Clifford Data Regression (CDR) | Error Mitigation | A learning-based EM technique that can, in some settings, improve the trainability of VQAs. | [41] |
| Variational Generative Optimization Network (VGON) | Classical ML Model | A deep generative model that finds optimal solutions to quantum problems, helping to avoid barren plateaus. | [42] |
| Exact Factorization (XF) Corrections | Theoretical Framework | Provides rigorous, derived correction terms (PQM, phase) for improved mixed quantum-classical dynamics. | [1] |
Frequently Asked Questions
What is the primary source of force inconsistency in mixed quantum-classical dynamics? The most common source is the inaccurate computation of the energy gradient (force) on the quantum subsystem. This often stems from approximations made in hybrid algorithms, such as neglecting Pulay forces in variational quantum eigensolver (VQE) calculations, and errors in the quantum computer's measurement of observables that are fed back to the classical subsystem [22].
How does the choice of measurement protocol affect force consistency? Inconsistent measurement protocols for quantum variations can lead to violations of conservation laws [45]. For dynamics, only the two-times quantum observables protocol has been shown to satisfy fundamental criteria like conservation laws and no-signaling, which are essential for producing consistent forces between subsystems [45].
Can coherent quantum feedback help mitigate force inconsistencies? Yes, unlike measurement-based feedback which collapses the quantum state, coherent feedback maintains the full quantum state and implements deterministic, non-destructive operations. This can provide a more stable and consistent feedback signal to the classical subsystem, potentially reducing force inconsistencies [46].
Why do my classical trajectories become unstable despite an accurate ground-state VQE energy? An accurate energy does not guarantee an accurate energy gradient. The forces governing classical motion are derived from these gradients. Inaccuracies in the gradient calculation, such as the Hellmann-Feynman approximation without Pulay term corrections, can lead to unphysical forces and unstable trajectories [22].
Troubleshooting Guides
Issue: Inconsistent Forces Leading to Energy Drift
Problem Description: The total energy of the combined quantum-classical system is not conserved during dynamics simulation.
| Potential Cause | Diagnostic Steps | Recommended Solutions |
|---|---|---|
| Inaccurate Hellmann-Feynman Force [22] | Check if Pulay forces are neglected in the gradient calculation. Compare gradients from finite-difference methods against the analytic Hellmann-Feynman term. | Implement the full gradient expression including Pulay terms. If computationally prohibitive, validate that the Hellmann-Feynman approximation holds for your specific system and ansatz [22]. |
| Noisy Quantum Observable Measurement [27] | Monitor the standard error of the measured observables (e.g., energy gradients) over multiple quantum circuit executions. | Increase the number of measurement shots (repetitions) for the observable to reduce statistical error. Use error mitigation techniques to improve measurement fidelity [27]. |
| Violation of Conservation Laws by Measurement Protocol [45] | Verify if the protocol used for measuring energy changes (a source of force) adheres to conservation laws. The two-point measurement (TPM) is known to fail in this regard. | Adopt the two-times quantum observables (OBS) protocol to characterize the variation of physical quantities, as it is proven to satisfy conservation laws [45]. |
Issue: Feedback Loop Instability
Problem Description: Small errors in the feedback between the quantum and classical subsystems amplify over time, causing the simulation to diverge.
| Potential Cause | Diagnostic Steps | Recommended Solutions |
|---|---|---|
| Time Delay in Feedback Loop [47] | Profile the computation time for the entire feedback cycle (classical position update -> Hamiltonian update -> quantum evolution -> observable measurement). | Optimize classical co-processing and use faster quantum controllers. Implement real-time estimation and compensation for fluctuating parameters, as demonstrated in spin qubit control [47]. |
| Error Propagation in Variational Optimization [27] | Track the infidelity of the quantum state at each time step. Check for large jumps in variational parameters between steps. | Use robust time-dependent variational quantum propagation (TDVQP) algorithms. For longer simulations, consider resetting the variational optimization with a new reference state to avoid error accumulation [27]. |
| Incorrect Integrator Choice | Verify if the time step (∆t) is too large for the chosen classical integrator (e.g., Velocity Verlet). | Reduce the time step. For the TDVQP meta-algorithm, ensure the classical propagator and the Hamiltonian used for time evolution are compatible; higher-order integrators may be necessary [27]. |
Experimental Protocols for Consistent Dynamics
Protocol 1: Time-Dependent Variational Quantum Propagation (TDVQP)
This is a modular meta-algorithm for general mixed quantum-classical dynamics [27].
- Initialization: The algorithm begins with a parameterized quantum circuit
C(θ)initialized to a desired state|ψ₀〉, generated based on an initial HamiltonianH(𝐪₀). The parametersθ₀are found using a VQA [27]. - Observable Measurement: A set of observables
{Ô₀}are measured from|ψ₀〉to obtain expectation values{O₀}[27]. - Classical Subsystem Update: These observables are used to evolve the classical parameters from
𝐪₀to𝐪₁using a classical integrator (e.g., Velocity Verlet) [27]. - Quantum Subsystem Update: The quantum state is evolved from
|ψ₀〉to|ψ₁〉using the time evolution operatorexp(-iH₀Δt). In a physical implementation, the parametersθ₁for the state|ψ₁〉 ≈ C(θ₁)|0〉are found using a single step of a circuit compression algorithm like p-VQD [27]. - Iteration: Steps 2-4 are repeated, using the new classical parameters
𝐪ₙto generate the HamiltonianHₙfor each subsequent time step [27].
Feedback Loop in Mixed Quantum-Classical Dynamics
Protocol 2: Force Computation via VQE with Gradients
This protocol details how to compute consistent forces for the classical subsystem using a hybrid quantum-classical algorithm [22].
- Hamiltonian Formulation: The molecular Hamiltonian in second quantization is mapped to a qubit operator via a transformation (e.g., Jordan-Wigner). This yields a Hamiltonian
Hcomposed of a sum of Pauli stringsP̂α[22]. - Energy Calculation: The VQE algorithm is used to find the ground state energy
E = ⟨Ψ(θ)|H|Ψ(θ)⟩. The parametersθof the ansatzΨ(θ)are optimized on a classical computer, while the expectation values of each Pauli string⟨P̂α⟩are measured on a quantum device [22]. - Gradient (Force) Calculation: The force on a nucleus
Iin directionξis the negative gradient of the energy:-∂E/∂R_Iξ. The gradient is approximated by the Hellmann-Feynman term:-∂E/∂R_Iξ ≈ -⟨Ψ(θ)| ∂H/∂R_Iξ |Ψ(θ)⟩. The gradient of the Hamiltonian∂H/∂R_Iξis itself a sum of Pauli terms, whose expectation values are measured on the quantum computer [22]. - Note on Approximation: This protocol uses the Hellmann-Feynman approximation, which disregards Pulay terms (
∂⟨Ψ(θ)|/∂R_Iξ H Ψ(θ)⟩ + c.c.). This approximation is common but can be a source of force inaccuracy and must be validated [22].
The Scientist's Toolkit: Research Reagent Solutions
| Item/Technique | Function in Ensuring Force Consistency |
|---|---|
| Two-Times Observables (OBS) Protocol [45] | Provides a theoretically consistent method for measuring energy changes in the quantum subsystem, respecting conservation laws and preventing a major source of force inconsistency. |
| Variational Quantum Eigensolver (VQE) [22] | A hybrid algorithm used to compute the ground and excited state energies of the quantum subsystem, which are essential for calculating the forces on classical particles. |
| Real-Time Quantum Controllers [47] | Hardware (e.g., OPX+) that enables rapid estimation of fluctuating parameters and real-time feedback, reducing time delays in the feedback loop that can cause instability. |
| Time-Dependent Variational Principle [27] | A class of algorithms (e.g., p-VQD, TDVQP) for propagating the quantum state in time, which helps manage error propagation and maintain consistency between classical and quantum states. |
| Coherent Feedback Control [46] | A feedback method that avoids wavefunction collapse, providing a more deterministic and potentially more consistent feedback signal compared to measurement-based feedback. |
Strategies for Initializing Classical Variables in the Single-Quantum Limit
In mixed quantum-classical (MQC) dynamics, the choice of initial conditions for classical variables is not merely a technical detail but a critical factor determining the stability and physical accuracy of simulations. Within the single-quantum limit, where quantum effects are pronounced and system sizes are minimal, improper initialization can lead to severe issues such as the barren plateau problem in variational quantum algorithms (VQAs), artificial coherence, and unphysical energy flow. This technical support guide addresses the specific challenges researchers face and provides actionable troubleshooting protocols to ensure reliable simulations.
Troubleshooting Guides
Guide 1: Addressing Barren Plateaus in Variational Quantum Algorithms
Problem Statement: Exponentially small gradients (barren plateaus) are encountered during the optimization of parameterized quantum circuits (PQCs), halting convergence.
Diagnosis and Solution: The barren plateau phenomenon, where gradients vanish exponentially with system size, is often linked to random initialization that explores unproductive regions of the Hilbert space. Mitigation strategies focus on informed initialization.
Recommended Initialization Strategy: Identity-Block Initialization This strategy initializes the PQC as a sequence of shallow blocks that each evaluate to the identity operation, limiting the effective depth at the start of training and preventing the circuit from being immediately trapped in a barren plateau [48].
Experimental Protocol:
- Circuit Construction: Design your parameterized quantum circuit in layers.
- Parameter Assignment: For each layer, randomly select a subset of parameters. Set the remaining parameters to specific values that cause the individual layer to perform an identity transformation.
- Training Commencement: Begin the variational optimization process. The initial circuit's proximity to the identity operation helps maintain a measurable gradient signal.
- Validation: Monitor the gradient norms of the cost function in the initial training steps. Successful initialization should yield non-vanishing gradients.
Performance Comparison of Initialization Strategies: The following table summarizes the expected outcomes from different initialization approaches, based on empirical studies [49].
| Initialization Strategy | Expected Gradient Norm (Initial) | Convergence Likelihood | Key Pros & Cons |
|---|---|---|---|
| Random Gaussian | Exponentially small | Very Low | Pro: Simple. Con: High probability of barren plateaus. |
| Zero-Initialization | Small | Low | Pro: Simple. Con: Can lead to symmetric traps. |
| Small-Angle Init. | Moderate | Moderate | Pro: Keeps circuit near identity. Con: May limit expressivity. |
| Identity-Block Init. [48] | Large | High | Pro: Mitigates barren plateaus. Con: More complex setup. |
| Classical-Heuristic (Xavier/He) [49] | Moderate to Large | Moderate | Pro: Inspired by proven classical methods. Con: Benefits can be marginal and problem-dependent. |
Guide 2: Managing Coherence and Phase Evolution in MQC Dynamics
Problem Statement: Simulations exhibit unphysical long-range electronic coherence or incorrect phase evolution between Born-Oppenheimer states.
Diagnosis and Solution: In exact factorization-based MQC dynamics, the proper description of decoherence and phase evolution relies on including key correction terms in the equations of motion [1]. Neglecting these terms leads to the observed artifacts.
Recommended Initialization Strategy: Initialize with Projected Quantum Momentum and Phase Corrections The classical nuclear momenta should be initialized to account for the quantum momentum corrections derived from the exact factorization formalism [1].
Experimental Protocol:
- System Setup: Define the molecular system and its initial nuclear geometry.
- Force Calculation: Compute the initial classical force using the expression:
F_ν = -∇_ν ϵ~ + Ȧ_ν, which includes the time-dependent vector potentialA_ν[1]. - Momentum Initialization: Initialize the nuclear velocities/momenta based on the forces that incorporate the Projected Quantum Momentum (PQM) and the newly identified phase-correction term [1].
- Propagation: Propagate the dynamics using the corrected electronic time-dependent Schrödinger equation (TDSE):
iℏ d/dt |Φ_R(t)⟩ = (H_BO + H_ENC(1) + H_ENC(2) - ϵ~) |Φ_R(t)⟩whereH_ENC(1)contains the PQM correction [1]. - Validation: Benchmark the dynamics against known results for models like the double-arch geometry (DAG) or two-dimensional nonseparable (2DNS) systems to verify accurate capture of nonadiabatic features like Stückelberg oscillations [1].
Guide 3: Optimizing for Remote State Preparation and Multiplexing
Problem Statement: Low rates and high infidelities in quantum network protocols like remote state preparation (RSP) and entanglement generation (EG).
Diagnosis and Solution: In asymmetric quantum network nodes, standard classical multiplexing fails to fully utilize available resources. Initialization strategies should account for novel quantum techniques like quantum multiplexing and multi-server multiplexing [50].
Recommended Initialization Strategy: Initialize for Coherent State Splitting For a weak coherent pulse (WCP) client, initialize the system to enable the splitting of a single quantum state across multiple spatial modes. This is represented by the transformation:
ξ|∅⟩ + √(1-ξ²)|1⟩ → ξ|∅⟩ + Σ_k √[(1-ξ²)/M] |1_k⟩[50].Experimental Protocol:
- Resource Assessment: Determine the number of available spatial modes
Mand the asymmetry between node memories. - State Preparation: Initialize the photonic state in a superposition of vacuum and a single photon.
- Linear Optics Setup: Configure a beam splitter array to implement the transformation that splits the single-photon component coherently across
Mmodes. - Protocol Execution: Run the RSP or EG protocol (e.g., single-click protocol). The multiplexed approach interacts with multiple quantum memories simultaneously, improving the overall rate-fidelity tradeoff [50].
- Metric Calculation: Calculate the multiplexing improvement factor
m_s = R_s|F(M) / R_s|F(1)to quantify the performance gain over the un-multiplexed case [50].
- Resource Assessment: Determine the number of available spatial modes
Frequently Asked Questions (FAQs)
FAQ 1: What is the single most critical factor when initializing classical variables for VQAs? The most critical factor is avoiding the random exploration of the entire Hilbert space. Initialization strategies that start the parameterized quantum circuit close to the identity operation (e.g., small-angle or identity-block initialization) have proven effective in mitigating the barren plateau problem and preserving gradient signal [48] [49].
FAQ 2: Can successful initialization strategies from classical deep learning be directly applied to quantum circuits? Direct application provides only marginal benefits. While classical methods like Xavier and He initialization are grounded in maintaining signal variance across layers, their translation to quantum circuits is non-trivial. Heuristic adaptations using global or chunk-based layerwise approaches have been tested, but their overall effectiveness is limited and problem-dependent, indicating a need for more quantum-aware initialization theories [49].
FAQ 3: How does initialization impact hybrid quantum-classical algorithms beyond VQE? Initialization is crucial for performance across the hybrid computing stack. For instance:
- In quantum neural networks (QNNs) for solving differential equations, proper initialization of feature map parameters is key to convergence and accuracy [51].
- In quantum-classical optimization (e.g., QAOA), initializing with classically generated pure Gibbs states has been shown to significantly improve solution quality compared to standard states [52].
- In quantum internet applications, initializing systems for quantum multiplexing directly enhances communication rates in asymmetric nodes [50].
FAQ 4: What is a "pure Gibbs state" and why is it a good initial state for QAOA? A pure Gibbs state is a quantum state that approximates the thermal Gibbs distribution while remaining a pure state, rather than a mixed state. It is characterized by a specific energy and coherence entropy. Research shows that initializing the Quantum Approximate Optimization Algorithm (QAOA) with low-temperature pure Gibbs states is a highly effective strategy, as these states lie on the "Boltzmann boundary" in the energy-entropy diagram and lead to higher-quality solutions after optimization [52].
Workflow Visualization
The following diagram illustrates a recommended decision workflow for selecting an initialization strategy, based on the primary simulation task.
Research Reagent Solutions
The table below lists key computational "reagents" — algorithms, ansätze, and protocols — essential for experiments in mixed quantum-classical dynamics.
| Research Reagent | Function & Application |
|---|---|
| Identity-Block Initialization [48] | Mitigates barren plateaus in VQEs and QNNs by initializing circuits as sequences of near-identity blocks. |
| Variational Quantum Eigensolver (VQE) [53] | A hybrid algorithm used to find ground/excited states of molecules; provides energies, gradients, and NACs for MQC dynamics. |
| Quantum Subspace Expansion (QSE) | Computes excited states and nonadiabatic coupling vectors from a ground state, used in dynamics simulations [53]. |
| Single-Click Protocol [50] | A protocol for remote state preparation or entanglement generation in quantum networks, amenable to quantum multiplexing. |
| Exact Factorization (XF) Framework [1] | Provides a rigorous foundation for deriving MQC equations of motion that include electron-nuclear correlation effects. |
| Pure Gibbs State [52] | An effective initial state for QAOA, found to improve solution quality by providing a favorable starting point on the energy-entropy landscape. |
Optimizing Ansatz Selection and Measurement Strategies for Noisy Hardware
Frequently Asked Questions
What is the most significant bottleneck for VQE on noisy hardware? Finite-shot sampling noise is a critical bottleneck. It distorts the cost landscape, creates false variational minima, and can lead to a statistical bias known as the "winner's curse," where the lowest observed energy is biased downward relative to the true value [54].
Which classical optimizers are most resilient to noise in VQE? Adaptive metaheuristic optimizers, specifically CMA-ES and iL-SHADE, have been identified as the most effective and resilient strategies. Gradient-based methods (e.g., SLSQP, BFGS) often struggle, as they can diverge or stagnate in noisy regimes [54].
How can I mitigate readout errors when estimating molecular energies? Using Quantum Detector Tomography (QDT) alongside informationally complete (IC) measurements can significantly reduce estimation bias. One study demonstrated a reduction in measurement errors by an order of magnitude, from 1-5% to 0.16%, for a molecular energy estimation on IBM hardware [24].
Are there optimizers designed specifically for physically-motivated ansätze? Yes, ExcitationSolve is a quantum-aware, gradient-free optimizer designed for ansätze composed of excitation operators (e.g., those in UCCSD). It determines the global optimum for each parameter and has been shown to converge faster and achieve chemical accuracy even in the presence of real hardware noise [55].
What is a fundamental limitation of noisy quantum computers? Noise places a fundamental constraint on achieving quantum advantage. For tasks like sampling from a quantum circuit's output, too much noise can allow the computation to be simulated efficiently by classical algorithms, restricting quantum advantage to a "Goldilocks zone" of qubit count and noise level [56].
Troubleshooting Guides
Problem: Optimizer Stagnation or Divergence
Potential Cause: The classical optimizer is being misled by a noisy energy landscape, characterized by false minima and barren plateaus [54]. Solutions:
- Switch Optimizer Class: Replace gradient-based methods (like BFGS or SLSQP) with adaptive metaheuristics like CMA-ES or iL-SHADE [54].
- Use Quantum-Aware Optimizers: For ansätze with excitation operators, implement ExcitationSolve. For ansätze with Pauli rotation gates, consider Rotosolve [55].
- Correct for Bias: When using population-based optimizers, track the population mean energy instead of the best individual's energy to correct for the "winner's curse" bias [54].
Problem: High Measurement Error and Shot Overhead
Potential Cause: Readout errors and a limited number of measurement shots (N_shots) lead to imprecise expectation values [54] [24].
Solutions:
- Implement Advanced Measurement Techniques: Use informationally complete (IC) measurements paired with Quantum Detector Tomography (QDT) to characterize and mitigate readout errors [24].
- Apply Shot-Frugal Strategies: Leverage locally biased random measurements to prioritize measurement settings that have a larger impact on the energy estimation, reducing the required number of shots [24].
- Mitigate Time-Dependent Noise: Use blended scheduling—interleaving circuits for QDT and energy estimation—to average out temporal noise fluctuations [24].
Problem: Physically Implausible Energy Results
Potential Cause: The variational ansatz does not conserve physical symmetries (e.g., particle number), or the noise has caused a violation of the variational principle [54] [55]. Solutions:
- Select a Physically-Motivated Ansatz: Use ansätze that respect the problem's physical symmetries, such as the Unitary Coupled Cluster (UCC) or the truncated Variational Hamiltonian Ansatz (tVHA) [54] [55].
- Co-Design Ansatz and Optimizer: Pair your physically-motivated ansatz with a compatible, noise-resilient optimizer like ExcitationSolve [54] [55].
Experimental Protocols & Data
Protocol 1: Benchmarking Optimizers Under Noise
This protocol outlines a method for comparing the performance of classical optimizers under simulated hardware noise, as derived from a large-scale benchmark [54].
- System Selection: Choose a test Hamiltonian (e.g., H₂, H₄ chain, or LiH).
- Ansatz Preparation: Select a variational ansatz (e.g., tVHA or a hardware-efficient ansatz).
- Noise Introduction: Simulate finite-shot noise by adding Gaussian noise to the exact energy expectation value:
ϵ_sampling ~ N(0, σ²/N_shots). - Optimizer Comparison: Run multiple optimization trials with different optimizers (e.g., CMA-ES, iL-SHADE, BFGS, SPSA, COBYLA) from the same set of initial parameters.
- Metrics: Record the final energy error (vs. FCI), number of iterations to convergence, and success rate.
Table 1: Optimizer Characteristics for Noisy VQE [54]
| Optimizer | Type | Key Characteristic | Performance under Noise |
|---|---|---|---|
| CMA-ES | Metaheuristic | Adaptive, population-based | Most effective and resilient |
| iL-SHADE | Metaheuristic | Improved adaptive differential evolution | Highly effective and resilient |
| SPSA | Gradient-based | Approximates gradient with few measurements | Moderate |
| COBYLA | Gradient-free | Linear approximation-based | Moderate |
| BFGS | Gradient-based | Uses exact gradient and Hessian approximation | Struggles, often diverges or stagnates |
| SLSQP | Gradient-based | Sequential quadratic programming | Struggles, often diverges or stagnates |
Protocol 2: High-Precision Measurement with Error Mitigation
This protocol details steps to achieve high-precision energy estimation on real, noisy hardware, as demonstrated for the BODIPY molecule [24].
- State Preparation: Prepare the target state on the quantum computer (e.g., a Hartree-Fock state).
- Design Measurements: Choose a set of informationally complete (IC) measurement settings. For efficiency, use a Hamiltonian-inspired, locally biased strategy.
- Parallel QDT: In the same execution batch, run circuits for Quantum Detector Tomography to characterize the readout noise model of the device.
- Blended Execution: Use a blended scheduling strategy to interleave the QDT circuits with the energy estimation circuits, mitigating the impact of time-dependent noise.
- Data Processing: Use the recorded QDT data to build an unbiased estimator for the energy, processing the IC measurement data accordingly.
Table 2: Error Mitigation Techniques and Their Impact [24]
| Technique | Function | Mitigated Error Source | Demonstrated Outcome |
|---|---|---|---|
| Quantum Detector Tomography (QDT) | Characterizes and corrects readout errors | Static readout noise | Reduced estimation bias |
| Locally Biased Measurements | Prioritizes informative measurement bases | Shot noise (finite statistics) | Reduced shot overhead |
| Blended Scheduling | Interleaves different circuit types | Time-dependent noise drift | Homogeneous noise across experiments |
This protocol describes how to use the ExcitationSolve algorithm to optimize parameters in an ansatz built from excitation operators [55].
- Parameter Selection: Isolate a single parameter
θ_jin the ansatzU(θ_j) = exp(-i θ_j G_j), where the generatorG_jsatisfiesG_j³ = G_j. - Energy Evaluation: For this parameter, evaluate the energy at a minimum of five different parameter values (e.g.,
θ_j,θ_j + π/2,θ_j + π,θ_j + 3π/2,θ_j + 2π). - Coefficient Fitting: Classically, fit the five energy values to the known analytical form of the energy landscape:
f(θ_j) = a₁ cos(θ_j) + a₂ cos(2θ_j) + b₁ sin(θ_j) + b₂ sin(2θ_j) + cto determine the coefficients. - Global Minimization: Find the global minimum of the fitted, noiseless analytic function using a classical method (e.g., a companion-matrix method).
- Parameter Update: Set
θ_jto the value that yields the global minimum. - Iterate: Sweep through all parameters in the ansatz and repeat until convergence.
The Scientist's Toolkit
Table 3: Essential Research Reagents & Resources [54] [2] [55]
| Item Name | Type | Function in Experiment |
|---|---|---|
| TEQUILA | Software Library | A quantum computing framework used for developing and running hybrid quantum-classical algorithms, such as VQE [2]. |
| ExcitationSolve | Optimization Algorithm | A quantum-aware, gradient-free optimizer for ansätze with excitation operators; finds the global optimum per parameter [55]. |
| SHARC | Software Package | A molecular dynamics program package used for non-adiabatic dynamics simulations, integrated with quantum computing frameworks [2]. |
| Informationally Complete (IC) Measurements | Measurement Strategy | A set of measurements that fully characterizes the quantum state, allowing estimation of multiple observables and enabling error mitigation via QDT [24]. |
| Hardware-Efficient Ansatz (HEA) | Quantum Circuit | A problem-agnostic ansatz designed for reduced depth on specific hardware; may not conserve physical symmetries [54]. |
| Unitary Coupled Cluster (UCCSD) | Quantum Circuit | A physically-motivated ansatz that conserves physical symmetries like particle number, providing physically plausible states and energies [54] [55]. |
| CMA-ES | Classical Optimizer | An adaptive metaheuristic optimizer renowned for its resilience to noise in VQE optimization tasks [54]. |
Workflow Diagrams
VQE Optimization Under Noise
High-Precision Measurement Protocol
Benchmarking MQC Performance: Accuracy, Scalability, and Path to Quantum Advantage
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary mechanisms for azobenzene photoisomerization, and how do I determine which one is operative in my system?
The photoisomerization of azobenzenes primarily proceeds via two competing mechanisms: rotation around the N=N double bond and inversion (concerted inversion) at one of the nitrogen atoms [57] [58]. The dominant pathway depends on the specific azobenzene derivative and the excited state that is initially populated.
- Rotation Mechanism: This pathway involves the torsional motion around the N=N double bond, similar to the isomerization of stilbene. It is typically associated with the excitation to the
S₂(π,π*)state and involves a significant change in the CNNC dihedral angle [57] [58]. - Inversion Mechanism: This pathway involves a change in the CNN bond angle where one of the nitrogen atoms undergoes a inversion motion, flipping the configuration. It is typically favored upon excitation to the
S₁(n,π*)state [57] [58].
Troubleshooting Tip: If your experiments show a violation of Kasha's rule (i.e., the quantum yield depends on the excitation wavelength), it is a strong indicator that multiple isomerization mechanisms are active. For instance, a higher quantum yield upon S₁(n,π*) excitation suggests a rotational pathway is favorable, while excitation to S₂(π,π*) may engage the inversion mechanism [57].
FAQ 2: Why is the quantum yield for my azobenzene derivative lower than expected, and how can I improve it?
A low photoisomerization quantum yield (Φ) can stem from several factors. The table below summarizes common causes and potential solutions based on recent case studies.
Table: Troubleshooting Low Photoisomerization Quantum Yields
| Cause | Description | Validation & Solution |
|---|---|---|
| Incorrect Excitation Wavelength | The quantum yield is highly dependent on the excited state populated. | Confirm the absorption maxima for both π→π* and n→π* transitions for your specific compound. Perform action spectroscopy to determine the optimal wavelength for the desired isomerization [58] [59]. |
| Environmental Effects | The surrounding environment (e.g., solvent, protein pocket) can restrict molecular motion. | The quantum yield of azo-escitalopram was found to be environment-dependent [59]. Compare results in different solvents (e.g., polar vs. non-polar) or in the gas phase to isolate environmental impact. |
| Competing Relaxation Pathways | The molecule may relax through pathways that do not lead to isomerization. | Use ultrafast spectroscopy to measure excited-state lifetimes. Long lifetimes, as seen in azo-escitalopram, can suggest slower torsional motion and lower yields [59]. Computational studies can map competing relaxation pathways to conical intersections [57]. |
| Molecular Design | The substituents on the azobenzene core can dictate the preferred mechanism and efficiency. | Electron-donor and acceptor groups can create "pseudo-stilbene" type azobenzenes with red-shifted absorption but potentially different isomerization dynamics [58]. Re-evaluate the substituent effects on your derivative. |
FAQ 3: My non-adiabatic dynamics simulations are not reproducing experimental quantum yields. What critical steps am I missing?
Reproducing experimental observables like quantum yields requires careful attention to the following in your simulation protocol:
- Accurate Electronic Structure Theory: Use high-level ab initio methods like CASSCF to correctly describe excited-state potential energy surfaces (PES), including key features like conical intersections (CI) and minimum energy conical intersections (MECI) [57] [60]. Semiempirical methods (e.g., FOMO-CI) can be used for larger systems but must be parameterized carefully [59].
- Sufficient Statistical Sampling: Quantum yields are statistical in nature. For Non-adiabatic Molecular Dynamics (NAMD) simulations, you must run a large number of trajectories (e.g., >100) to obtain statistically meaningful results [61].
- Inclusion of Solvent Effects: The solvent environment can significantly alter dynamics. Implement a Quantum Mechanics/Molecular Mechanics (QM/MM) framework to include explicit solvent molecules in your simulation [59].
- Check for Multiple Isomerization Pathways: Systems like azo-escitalopram can form multiple cis-isomers [59]. Ensure your simulation and analysis can account for and distinguish between all possible photoproducts.
Troubleshooting Guides
Issue: Inconsistent or Irreproducible Photoisomerization Results
Potential Cause 1: Unstable Light Source or Incorrect Wavelength
- Validation Protocol: Use a calibrated power meter at the sample position to verify light intensity before each experiment. Ensure the bandwidth of your light source (e.g., laser, LED) matches the target absorption band (
n,π*vsπ,π*). - Solution: Regularly maintain and calibrate your light source. Use monochromators or specific bandpass filters to ensure spectral purity.
Potential Cause 2: Failure to Reach a Photostationary State (PSS)
- Validation Protocol: Monitor the reaction progress using UV-Vis spectroscopy or
¹H NMRover time until no further spectral changes are observed. The composition of the PSS is wavelength-dependent [58]. - Solution: Irradiate the sample for a sufficient duration to reach the PSS. The time required can be determined empirically. For quantification, use
¹H NMRto measure the precise isomeric ratio at the PSS [60].
Potential Cause 3: Sample Decomposition or Fatigue
- Validation Protocol: After irradiation, analyze the sample using techniques like HPLC or mass spectrometry to check for side products or decomposition.
- Solution: Azobenzenes can undergo fatigue over many cycles [58]. Use degassed solvents to minimize oxidative degradation and avoid excessively long irradiation times with high-power sources.
Issue: Challenges in Simulating Electronic Relaxation Pathways
Potential Cause 1: Inadequate Conical Intersection Sampling
- Validation Protocol: Optimize Minimum Energy Conical Intersections (MECIs) using methods like CASSCF. Validate that the geometries and energies of these MECIs are consistent with the reaction path [57] [60].
- Solution: When running dynamics, ensure trajectories pass through or near these validated MECIs. The "growing string" method can be used to find multiconfigurational reaction paths to MECIs [60].
Potential Cause 2: Poor Treatment of Solvent in QM/MM Setup
- Validation Protocol: Run a control simulation varying the number of explicit solvent molecules or the size of the QM region to ensure your results are converged.
- Solution: As demonstrated in the azo-escitalopram study [59], include enough explicit solvent molecules in the QM region to capture essential solute-solvent interactions (e.g., hydrogen bonding).
Potential Cause 3: High Error Rates in Quantum Hardware for Hybrid Simulations
- Validation Protocol: When using variational quantum eigensolvers (VQE), benchmark the calculated energies against classical results for small, known systems.
- Solution: Mitigate hardware noise by using error correction codes or advanced algorithms. For example, the
pUCCD-DNNapproach uses deep neural networks to optimize wavefunctions, reducing the number of quantum hardware calls and making the process more robust to noise [62].
Experimental Protocols & Data
Protocol 1: Ultrafast Spectroscopy to Probe Isomerization Dynamics
This protocol outlines the use of transient absorption spectroscopy to track the real-time dynamics of photoisomerization [60].
- Sample Preparation: Prepare a dilute solution of the photoswitch (e.g.,
~0.1 ODat the excitation wavelength) in an appropriate solvent. Degas the solution to prevent photo-oxidation. - Pump-Probe Setup: Use a femtosecond laser system. The pump pulse is tuned to the desired absorption band (
S₂(π,π*)orS₁(n,π*)). A white-light continuum serves as the probe pulse across a broad spectral range. - Data Collection: Record differential absorption (
ΔA) spectra at various time delays between the pump and probe pulses (from femtoseconds to nanoseconds). - Data Analysis: Global fitting and target analysis are used to extract species-associated difference spectra (SADS) and the corresponding time constants for processes like internal conversion, vibrational cooling, and isomerization.
Protocol 2: Quantum Chemical Calculation of Reaction Pathways
This protocol describes a computational methodology for mapping the photoisomerization mechanism [57] [60].
- Geometry Optimizations:
- Use the CASSCF method with an active space appropriate for the system (e.g., CAS(10,8) for azobenzene) to optimize the ground-state structures of the
transandcisisomers [57]. - Locate the transition state (TS) on the ground-state surface connecting the two isomers.
- Use the CASSCF method with an active space appropriate for the system (e.g., CAS(10,8) for azobenzene) to optimize the ground-state structures of the
- Excited-State Mapping:
- Optimize the minimum energy structures for the low-lying excited states (
S₁andS₂). - Perform relaxed potential energy surface (PES) scans along key reaction coordinates (CNNC dihedral for rotation; CNN angle for inversion).
- Optimize the minimum energy structures for the low-lying excited states (
- Locating Conical Intersections:
- Validation: Compare calculated energies (reaction barriers, excitation energies) and geometries with available experimental data.
Table: Key Quantitative Data from Photoisomerization Studies
| System | Excitation | Quantum Yield (Φ) | Lifetime (τ) | Key Findings | Source |
|---|---|---|---|---|---|
| Azobenzene | S₁(n,π*) |
0.20–0.36 |
- | Rotational mechanism favored. | [57] |
| Azobenzene | S₂(π,π*) |
0.09–0.20 |
110–170 fs (S₂ lifetime) |
Can decay via rotation or concerted-inversion. Fast relaxation to S₁. | [57] |
| 2PyMIG | S₂(π,π*) |
- | Picoseconds | Isomerization via small barrier (4.3-6.5 kcal/mol) on S₂ PES before reaching S₂/S₀ MECI. |
[60] |
| Azo-escitalopram | n→π* |
Higher than π→π* |
Longer than azobenzene | Quantum yield is wavelength and environment dependent. Two distinct cis-isomers formed. | [59] |
| Isotopically Chiral Motor 2 | S₂(π,π*) |
High (similar to chem. chiral) | - | Demonstrated potential for fast, unidirectional rotary motion. | [61] |
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Computational and Experimental Tools
| Item / Reagent | Function / Application | Example from Literature |
|---|---|---|
| CASSCF Method | High-level ab initio quantum chemistry method for accurately modeling photochemical reactions and optimizing critical structures like conical intersections. | Used to optimize conical intersections and map the PES for azobenzene photoisomerization [57]. |
| Trajectory Surface Hopping (TSH) | A mixed quantum-classical dynamics method for simulating non-adiabatic processes like photoisomerization, where trajectories can "hop" between electronic states. | Implemented with FOMO-CI to study the photoisomerization dynamics of azo-escitalopram in gas phase and water [59]. |
| Quantum Mechanics/Molecular Mechanics (QM/MM) | A hybrid computational approach that models the core region of interest with quantum mechanics while treating the surrounding environment with molecular mechanics. | Used to include explicit water molecules in the dynamics simulations of azo-escitalopram [59]. |
| Variational Quantum Eigensolver (VQE) | A hybrid quantum-classical algorithm used to find ground or excited states of molecular systems, suitable for current noisy quantum hardware. | Paired with a deep neural network (pUCCD-DNN) to improve the accuracy of molecular energy calculations and model reactions like isomerization [62]. |
| Ultrafast Laser Spectroscopy | Experimental technique (e.g., transient absorption) to probe photochemical reactions on femtosecond to nanosecond timescales, revealing intermediate states and kinetics. | Used to quantify the picosecond-scale photoisomerization and low fluorescence yields of the 2PyMIG switch [60]. |
Workflow and Pathway Visualizations
Diagram: Azobenzene Photoisomerization Workflow
The diagram below illustrates the key steps for a combined computational and experimental study of azobenzene photoisomerization, highlighting the validation feedback loop.
Diagram: Competing Photoisomerization Pathways
This diagram outlines the two primary mechanistic pathways for azobenzene photoisomerization and their connections to different electronic states.
Frequently Asked Questions
What are the most common failure modes when mixed quantum-classical (MQC) methods fail to reproduce Stückelberg oscillations? The failure to capture Stückelberg oscillations typically stems from two primary issues:
- Inadequate Treatment of Electronic Phase Evolution: Many standard MQC methods, including conventional Ehrenfest dynamics and surface hopping, lack a rigorous mechanism for the proper phase evolution of the electronic coefficients. This phase is crucial for the constructive and destructive interference that forms Stückelberg patterns. Without specific corrections, these methods cannot reproduce the oscillations accurately [1].
- Missing Electron-Nuclear Correlation (ENC) Effects: The absence of terms accounting for electron-nuclear correlation in the equations of motion leads to an incomplete physical picture. Newly derived MQC methods that incorporate Projected Quantum Momentum (PQM) and a previously unidentified phase-correction term within the Exact Factorization framework have demonstrated a balanced and rigorous description of both decoherence and phase dynamics, which is essential for capturing Stückelberg oscillations [1].
Which experimental platforms provide the most reliable benchmarks for Stückelberg interference? Programmable Rydberg atom arrays and superconducting fluxonium qubits are leading platforms for benchmarking due to their high controllability and clear readout of interference patterns.
- Programmable Rydberg Arrays: Experiments on arrays of up to 100 atoms have demonstrated robust many-body Stückelberg interference, with visibility exceeding 70% and excitation suppression down to 1%. The system's Hamiltonian is well-known, and driving protocols can modulate both detuning and Rabi frequency, providing a rich testbed for dynamics [63].
- Fluxonium Qubits: The multi-level structure of fluxonium circuits under strong periodic driving produces characteristic LZS interference spectra. The resonances in these spectra originate from constructive interference favoring transitions to higher levels, providing a clear, measurable signature for validating simulations [64].
How can I determine if my MQC simulation has successfully captured coherent dynamics? Successful capture of coherence is indicated by the emergence of specific quantum phenomena in your simulation results. Key indicators include:
- Observed Stückelberg Oscillations: The appearance of oscillatory patterns in population dynamics or time-averaged observables (like Rydberg excitation density) as a function of a driving parameter (e.g., frequency or amplitude) is a direct signature of coherent interference [63] [65].
- Recoherence and Population Cycling: Instead of a monotonic decay, you should observe oscillatory recoherence and population cycling between states, indicating the system is undergoing coherent dynamics rather than simply relaxing [65].
- Quantitative Benchmarking: Compare your results against exact grid-based quantum dynamics simulations on low-dimensional models or the experimental benchmarks mentioned above [66].
Our hybrid quantum-classical algorithm suffers from error propagation. How can this be mitigated? In hybrid algorithms like Time-Dependent Variational Quantum Propagation (TDVQP), errors propagate between the quantum and classical subsystems. Coherent errors in the quantum computer's wavefunction representation affect the measured observables. These inaccurate forces then lead to incorrect evolution of the classical parameters, which in turn define an erroneous Hamiltonian for the next quantum step, creating a feedback loop of inaccuracies [27]. Mitigation strategies include:
- Using highly expressive quantum circuit ansatze to better represent the quantum state.
- Employing shot-efficient optimizers to reduce measurement noise.
- Implementing higher-order classical integrators that may be more robust to force inaccuracies [27].
Troubleshooting Guides
Problem: Missing Stückelberg Oscillations in Surface Hopping Simulations
Issue: Your trajectory surface hopping simulation results show monotonic population decay instead of the expected oscillatory Stückelberg interference pattern.
Diagnosis: This is a classic symptom of the lack of a rigorous mechanism for electronic phase evolution and decoherence in standard mixed quantum-classical methods.
Solution:
- Implement Corrected Equations of Motion: Move beyond basic surface hopping or Ehrenfest dynamics. Adopt newly developed MQC equations derived from the Exact Factorization framework.
- Incorporate Key Terms: Ensure your solver includes both the Projected Quantum Momentum (PQM) correction and the additional phase-correction term. These corrections, which appear at the order of ℏ, are essential for a unified and rigorous account of electronic coherence and phase evolution [1].
- Validation: Test your updated implementation on benchmark models like the double-arch geometry (DAG) or two-dimensional nonseparable (2DNS) models, which are known to exhibit Stückelberg oscillations and other nonadiabatic features [1].
Problem: Incoherent Dynamics in Hybrid Quantum-Computing Algorithms
Issue: Your hybrid quantum-classical algorithm, where the quantum part runs on a quantum computer, produces results that deviate from exact benchmarks and show suppressed coherence.
Diagnosis: The inherent noise and errors in near-term quantum hardware (NISQ devices) destroy the delicate phase coherence necessary for effects like Stückelberg oscillations.
Solution:
- Algorithmic Structure: Employ a structured algorithm like the Time-Dependent Variational Quantum Propagation (TDVQP). This is a modular meta-algorithm where the quantum subsystem (e.g., electrons in a diabatic basis) is evolved on the quantum processor, and the classical subsystem (e.g., nuclei) is evolved based on measured observables [27].
- Circuit Design:
- Ansatz Selection: Use a sufficiently expressive, potentially adaptive, parameterized quantum circuit ansatz (\hat{C}(\overrightarrow{\theta})) to represent the quantum state [27].
- Time Evolution: Implement the time evolution using a Trotterized form of the time evolution operator, (\exp(-i{\hat{H}}_{0}\Delta t)), as done in the p-VQD algorithm [27].
- Error Management: Focus on short-time evolution where the cumulative errors are smaller. The TDVQP algorithm has been shown to perform well for short-time evolutions and retain qualitative results for longer times [27].
Benchmarking Data & Experimental Protocols
Quantitative Benchmarks for Method Validation
The following table summarizes key metrics from experimental and theoretical studies that can be used as benchmarks for validating MQC dynamics simulations.
Table 1: Experimental Benchmarks for Stückelberg Oscillations
| System | Observable | Target Performance | Key Parameter Ranges | Citation |
|---|---|---|---|---|
| Programmable Rydberg Array | Rydberg excitation density (n(T)) after one drive cycle | Interference visibility >70%, excitation suppression to ~1% | (\Omega0 = 2\ \text{rad/μs}), (\Delta0 = 20\ \text{rad/μs}), atom spacing (4.7\ \mu\text{m}) [63] | [63] |
| 1D Atom Chain (100 atoms) | Stückelberg interference pattern (excitation vs. drive frequency) | Pronounced "bright" (excitation) and "dark" (freezing) fringes | Drive frequency (\omega) is the varied parameter [63] | [63] |
| Fluxonium Qubit | Time-averaged level population under large-amplitude drive | Resonance patterns indicating transitions to higher levels | Drive amplitude (A), flux detuning (\delta_0) [64] | [64] |
Table 2: MQC Method Performance on Model Systems
| Method / Framework | Captures Stückelberg Oscillations? | Key Strengths | Key Limitations / Requirements |
|---|---|---|---|
| Exact Factorization with PQM & Phase Corrections | Yes [1] | Rigorous foundation; unified treatment of decoherence and phase | Derived complexity; requires implementation of new force terms [1] |
| Grid-Based Split-Operator (Full Quantum) | Yes (exactly) [66] | Exact treatment; includes all interference and geometric phases | Scalability limited to low-dimensional systems (2-4D) [66] |
| Gaussian Wave Packets (e.g., MCA) | Yes [66] | Good balance of accuracy and scalability for moderate dimensions | Accuracy depends on the number of basis functions [66] |
| Hybrid Quantum Computing (TDVQP) | Qualitative for short times [27] | Potential for quantum advantage on larger systems | Susceptible to hardware noise and error propagation [27] |
| Standard Surface Hopping | Typically No [1] | High scalability for large systems | Lacks rigorous phase evolution and decoherence treatment [1] [66] |
Detailed Experimental Protocol: Many-Body Stückelberg Interference in Rydberg Arrays
This protocol is based on the experiment documented in [63] and serves as an excellent benchmark.
1. System Setup:
- Platform: QuEra's Aquila programmable neutral-atom quantum simulator or an equivalent system.
- Geometry: Arrange atoms in a one-dimensional "snake-like" chain or a two-dimensional lattice. A 100-atom chain is a standard test.
- Initialization: Prepare the system in the collective vacuum state (|v\rangle), where all atoms are in the ground state (|g\rangle).
- Hamiltonian: The system is governed by the Rydberg atom Hamiltonian:
(
H(t) = -\sumi \Delta(t) ni + \sumi \frac{\Omega(t)}{2} \sigmai^x + \sum{i
2. Drive Protocols: Choose one of two modulation protocols to apply after initialization.
- Single-Frequency Protocol:
- (\Delta(t) = \Delta0 \cos(\omega t))
- (\Omega(t) = \Omega0) (constant)
- Bi-Frequency Protocol (Enhanced Suppression):
- (\Delta(t) = \Delta0 \cos(\omega t))
- (\Omega(t) = \frac{\Omega0}{2}[1 + \cos(r\omega t)]), where (r=2) introduces a second harmonic.
3. Measurement:
- After applying the drive for one full cycle (time (T)), measure the Rydberg excitation density (n(T)).
- Repeat this measurement while systematically varying the drive frequency (\omega) (and potentially amplitude (\Delta_0)) to map out the interference pattern.
4. Expected Outcome: A successful experiment or simulation will show clear oscillations in (n(T)) as a function of (\omega). These are the Stückelberg oscillations, with peaks ("bright fringes") corresponding to high excitation and troughs ("dark fringes") corresponding to dynamical freezing of the vacuum state [63].
Workflow for Benchmarking a Mixed Quantum-Classical Method
The following diagram illustrates a general workflow for validating an MQC method against a known benchmark.
The Scientist's Toolkit: Essential Research Reagents & Solutions
This table lists key computational tools and theoretical concepts essential for research in this field.
Table 3: Key Research Reagents and Solutions
| Item / Concept | Function / Role | Example Implementation / Notes |
|---|---|---|
| Exact Factorization (XF) Framework | Provides a rigorous foundation for deriving improved mixed quantum-classical equations of motion that include full electron-nuclear correlation effects. | Used to derive PQM and phase corrections for accurate coherence and phase dynamics [1]. |
| SHARC-TEQUILA Integration | A software approach for nonadiabatic molecular dynamics where electronic properties are computed on a quantum computer via VQE, and nuclei are propagated classically. | Enables hybrid quantum-classical simulation of photoisomerization and electronic relaxation [2]. |
| Programmable Rydberg Simulator | A hardware platform for running experimental benchmarks on many-body quantum dynamics and interference. | QuEra's Aquila; allows testing of MQC methods against real, large-scale quantum systems [63]. |
| Time-Dependent Variational Quantum Propagation (TDVQP) | A NISQ-friendly algorithm for mixed quantum-classical dynamics. Offloads quantum subsystem evolution to a quantum computer. | A modular meta-algorithm; useful for testing on model systems like the Shin-Metiu model [27]. |
| Projected Quantum Momentum (PQM) & Phase Corrections | Specific additive terms in the electronic equation of motion. They are essential for capturing the correct phase evolution and decoherence needed for Stückelberg oscillations. | Must be implemented in MQC codes for accurate interference phenomena [1]. |
Frequently Asked Questions
Q1: My surface hopping simulations show unphysical long-lived electronic coherences. What is the cause and how can it be corrected?
This is a common issue caused by the lack of a proper decoherence mechanism in traditional surface hopping. In standard fewest-switches surface hopping (FSSH), the quantum coefficients can develop nonphysical coherences over large time scales because each trajectory evolves independently without accounting for wavepacket branching [67]. Two solutions are available:
- Implement artificial decoherence corrections such as the augmented FSSH (AFSSH) method, which enforces stochastic wave function collapse at a rate proportional to the difference in forces between the active surface and other surfaces [68].
- Switch to exact factorization (XF)-based methods where decoherence is naturally incorporated through the electron-nuclear correlation terms, particularly the quantum momentum, which couples electronic evolution to nuclear density variations [1] [69].
Q2: How do I handle "frustrated hops" where a trajectory lacks kinetic energy for a surface transition?
When a surface hop requires more energy than available in the velocity component along the nonadiabatic coupling vector [67]:
- The simplest approach is to ignore the hop and continue on the current surface.
- A better physical approach is to reverse the direction of the velocity component along the nonadiabatic coupling vector while remaining on the current surface.
- For XF-based methods, the use of coupled trajectories often reduces frustrated hops because the quantum momentum correction naturally accounts for nuclear delocalization effects [13].
Q3: What are the key differences in computational cost between exact factorization and traditional surface hopping?
Table: Computational Requirements Comparison
| Component | Traditional Surface Hopping | Exact Factorization Methods |
|---|---|---|
| Trajectory Propagation | Independent trajectories | Coupled or auxiliary trajectories [13] [14] |
| Electronic Structure | Energies, gradients, NAC vectors | Additional quantum momentum terms [1] |
| Memory Requirements | Lower per trajectory | Higher due to trajectory coupling [14] |
| Parallelization | Embarrassingly parallel | Requires communication between trajectories [13] |
Q4: Which approach provides better performance for multistate systems involving three or more electronic states?
For multistate dynamics, XF-based methods generally show superior performance, especially when multiple states are occupied simultaneously [14]. The electron-nuclear correlation terms in XF properly capture the coupling between all occupied states, whereas traditional surface hopping can struggle with coherent multistate interactions. Studies on the uracil radical cation through a three-state conical intersection demonstrate that XF methods maintain accuracy where traditional approaches may show deviations [14].
Troubleshooting Guides
Issue: Poor Energy Conservation in Dynamics Simulations
Symptoms: Total energy drifts more than 1% over simulation time, or trajectories show systematic energy gain/loss.
Solutions:
- For traditional surface hopping:
- Check the time step: Use a shorter electronic time step (
AIMD_SHORT_TIME_STEPin Q-Chem) while maintaining a reasonable nuclear time step [68]. - Verify the derivative coupling calculation: Ensure NAC vectors are numerically stable, especially near conical intersections.
- Check the time step: Use a shorter electronic time step (
- For exact factorization methods:
Recommended Diagnostic Table: Table: Energy Conservation Checks
| Checkpoint | Frequency | Acceptable Threshold |
|---|---|---|
| Total energy drift | Every 10 steps | < 0.1% fluctuation |
| Wave function norm | Every electronic step | > 0.999 trace of density matrix [68] |
| Quantum momentum stability | Every step (XF only) | Smooth spatial variation [1] |
Issue: Incorrect Branching Ratios in Photochemical Reactions
Symptoms: Predicted product ratios disagree with experimental measurements or exact quantum dynamics.
Solutions:
- Traditional surface hopping:
- Increase the number of trajectories, particularly for low-probability pathways.
- Implement energy-based decoherence corrections to improve branching ratios [67].
- Exact factorization approaches:
Branching Ratio Troubleshooting
Issue: Numerical Instabilities in Strong Field Dynamics
Symptoms: Wave function blow-up, unphysical population transfer, or code crashes in external fields.
Solutions:
- Both methods:
- Use the electronic representation best suited to the field strength: diabatic for strong fields, adiabatic for weak fields.
- For exact factorization specifically: The time-dependent vector potential
A_ν(R,t)must be carefully integrated to maintain numerical stability [13] [70]. - For traditional surface hopping: Consider Floquet or dressed states for periodically driven systems [13].
Experimental Protocol for Stability:
- Start with a minimal system (1D models if available)
- Test with zero field to establish baseline stability
- Gradually increase field strength while monitoring:
- Electronic population continuity
- Nuclear force smoothness
- Conservation laws
Research Reagent Solutions
Table: Essential Computational Tools for Mixed Quantum-Classical Dynamics
| Tool/Component | Function | Implementation Notes |
|---|---|---|
| Nonadiabatic Coupling Vectors | Govern transitions between electronic states | Critical for accurate surface hopping; requires analytic derivatives where possible [68] |
| Quantum Momentum Term | Encodes electron-nuclear correlation in XF | Calculated from nuclear density variations using coupled/auxiliary trajectories [1] |
| Decoherence Schemes | Correct unphysical long-lived coherences | Energy-based, force-based, or XF's natural decoherence through PQM [67] [1] |
| Coupled Trajectory Algorithms | Capture nuclear quantum effects in XF | CTMQC and CTSH provide more accuracy than independent trajectories [13] [14] |
| Hybrid Quantum-Classical Hardware | Compute electronic properties efficiently | Quantum computers with VQE algorithms for expensive electronic structure [2] [62] |
Method Selection Workflow
Experimental Protocol Standards
Standardized Testing for Method Validation
Before applying any method to new systems, validate against benchmark cases:
Protocol 1: Double-Arch Geometry (DAG) Model Test
- Purpose: Verify accurate treatment of Stückelberg oscillations and coherence effects [1]
- Procedure:
- Implement the 1D DAG Hamiltonian with two electronic states
- Initialize wavepacket on upper surface
- Propagate for 100 fs with 1000 trajectories
- Measure electronic populations and coherence
- Success criteria: Reproduction of characteristic oscillation patterns within 5% of exact quantum results
Protocol 2: Three-State Conical Intersection Test
- Purpose: Validate multistate dynamics capability [14]
- System: Uracil radical cation or model system
- Metrics:
- Population transfer rates between all state pairs
- Final product branching ratios
- Energy conservation throughout propagation
Implementation Checklist for Production Simulations
- Convergence test for trajectory count (start with 500-1000)
- Time step validation (electronic 0.1-0.5 au, nuclear 5-20× larger) [68]
- Decoherence scheme selection and parameter tuning
- Quantum momentum implementation (XF methods only) [1]
- Property output configuration (populations, coherences, geometries)
- Restart file configuration for long simulations [68]
Table: Recommended Methods by Application Scenario
| Application Scenario | Recommended Method | Key Parameters |
|---|---|---|
| Ground state dynamics | Traditional surface hopping | FSSH with decoherence |
| Strong field processes | Exact factorization (CTMQC) | Coupled trajectories, full PQM [13] |
| Multistate conical intersections | XF-based surface hopping | 3+ states, quantum momentum [14] |
| Large systems (>20 atoms) | Traditional surface hopping + decoherence | Independent trajectories |
| Quantum coherence studies | XF with phase correction | PQM + phase terms [1] |
Frequently Asked Questions
What are the primary sources of error in computational predictions of Gibbs free energy for solids? Benchmarking studies reveal that predictions of Gibbs free energy (G) for crystalline solids, whether from Machine Learning Interatomic Potentials (MLIPs) or Density Functional Theory (DFT), often lack the accuracy and precision required for robust thermodynamic modeling. Much of the calculated and experimental data itself can be a source of error, limiting reliable applications [71] [72].
Why might my deep-learning model for binding affinity perform well on benchmarks but fail in real-world drug design? A common issue is train-test data leakage. Performance is often inflated because of structural similarities between the standard training database (PDBbind) and the test benchmarks (CASF). When this leakage is eliminated using a curated dataset like PDBbind CleanSplit, the performance of many state-of-the-art models drops substantially, revealing poor generalization [73].
How can mixed quantum-classical dynamics simulations aid in drug discovery? Mixed quantum-classical (MQC) dynamics can model complex molecular systems by treating a reactive core (e.g., a ligand binding site) quantum mechanically and the surroundings classically. This is a promising method for understanding reaction mechanisms and binding events that are too complex for full quantum simulation, especially on near-term quantum computers [27].
What is the current state of quantum utility for simulating molecular dynamics? While quantum hardware is advancing, existing supercomputers still outperform quantum computers for most quantum-chemistry problems. Current quantum algorithms primarily study systems manageable on classical hardware. The focus is on developing hybrid algorithms, like the Time-Dependent Variational Quantum Propagation (TDVQP), for future advantage [27].
Troubleshooting Guides
Issue: Inaccurate Gibbs Free Energy Predictions
| Troubleshooting Step | Description & Rationale | Expected Outcome |
|---|---|---|
| 1. Intermethod Benchmarking | Compare your results against multiple methods (e.g., DFT, different MLIPs) and available experimental data [71] [72]. | Identifies systematic errors and establishes a performance baseline for your specific system. |
| 2. Validate Input Data | Scrutinize the accuracy and precision of the experimental or reference data used for training or validation [72]. | Ensures that model inaccuracies are not propagated from faulty reference data. |
| 3. Employ a Reaction Network | Feed your calculated Gibbs free energies into a reaction network (RN) to check for thermodynamic consistency [71]. | The RN can provide experimentally informed predictions and help identify outliers. |
Issue: Poor Generalization of Binding Affinity Models
| Troubleshooting Step | Description & Rationale | Expected Outcome |
|---|---|---|
| 1. Check for Data Leakage | Use a structure-based clustering algorithm to ensure no protein-ligand complexes in your training set are highly similar to those in your test set [73]. | Creates a strictly independent test set for a genuine evaluation of generalization capability. |
| 2. Reduce Training Set Redundancy | Filter out highly similar complexes from your training data to prevent the model from settling for memorization [73]. | Encourages the model to learn fundamental protein-ligand interactions rather than exploiting similarities. |
| 3. Use a Robust Model Architecture | Implement a model like the Graph neural network for Efficient Molecular Scoring (GEMS), which uses a sparse graph and transfer learning, and train it on a leakage-free dataset like PDBbind CleanSplit [73]. | Achieves high benchmark performance based on a genuine understanding of interactions, not data leakage. |
Issue: Error Propagation in Mixed Quantum-Classical Dynamics
| Troubleshooting Step | Description & Rationale | Expected Outcome |
|---|---|---|
| 1. Analyze Error Sources | Identify if inaccuracies stem from the quantum circuit compression, the time evolution approximation, or the classical propagator [27]. | Allows for targeted improvements in the specific component introducing the largest error. |
| 2. Short-Time Evolution Focus | Use the Time-Dependent Variational Quantum Propagation (TDVQP) algorithm for short-time evolutions, where it performs well [27]. | Provides accurate short-term dynamics, retaining qualitative results for longer simulations. |
| 3. Monitor Wavefunction Infidelity | The error in the quantum wavefunction can be represented as a superposition of the desired state and an error term. This infidelity directly propagates to observable measurements [27]. | Quantifying this infidelity helps understand the lower bound of error in calculated observables like forces. |
Data Presentation: Performance Benchmarks
Table 1: Performance of Gibbs Free Energy Prediction Methods This table summarizes the relative performance of different computational methods for predicting the Gibbs free energy of crystalline solids, as benchmarked against experimental data [71] [72].
| Method | Approximation | Performance & Key Findings |
|---|---|---|
| Machine Learning Interatomic Potentials (MLIPs) | Harmonic & Quasi-Harmonic | Shows promising performance but does not consistently outperform simpler methods when used within a Reaction Network [71]. |
| Density Functional Theory (DFT) | Harmonic & Quasi-Harmonic | A standard approach, but its accuracy can be limited for some solids compared to experimental data [71]. |
| Reaction Network (RN) | Experimentally Informed | When fed with calculated data, this method can provide satisfactory predictions of room-temperature Gibbs free energy of formation, sometimes outperforming direct MLIP predictions [71]. |
Table 2: Impact of Data Leakage on Binding Affinity Prediction Models (CASF Benchmark) This table illustrates how removing data leakage dramatically affects the reported performance of leading deep-learning scoring functions [73].
| Model | Trained on Original PDBbind | Trained on PDBbind CleanSplit (Reduced Leakage) | Implication |
|---|---|---|---|
| GenScore (State-of-the-Art) | Excellent benchmark performance | Performance drops markedly | Previous high performance was largely driven by data leakage and memorization [73]. |
| Pafnucy (State-of-the-Art) | Excellent benchmark performance | Performance drops markedly | Previous high performance was largely driven by data leakage and memorization [73]. |
| GEMS (Graph Neural Network) | Not Applicable | Maintains high, state-of-the-art performance | Demonstrates robust generalization to strictly independent test data [73]. |
Experimental Protocols
Protocol 1: Creating a Clean Dataset for Binding Affinity Prediction Objective: To generate a training dataset (e.g., PDBbind CleanSplit) free of train-test data leakage and redundancy to enable genuine model generalization [73].
- Similarity Calculation: For all protein-ligand complexes in the training set (PDBbind) and test set (CASF), compute a multi-modal similarity score. This combines:
- Protein Similarity: Using TM-scores.
- Ligand Similarity: Using Tanimoto scores (based on molecular fingerprints).
- Binding Conformation Similarity: Using pocket-aligned ligand root-mean-square deviation (RMSD).
- Remove Test Leakage: Identify and exclude all training complexes that are highly similar to any test complex based on the combined metrics. Also, remove any training complexes with ligands identical (Tanimoto > 0.9) to test ligands.
- Reduce Redundancy: Within the training set, identify clusters of highly similar complexes. Iteratively remove complexes from these clusters until the most striking redundancies are resolved, creating a more diverse training set.
Protocol 2: Implementing Mixed Quantum-Classical (MQC) Dynamics with TDVQP Objective: To simulate the non-adiabatic dynamics of a molecular system by coupling a quantum subsystem (e.g., electrons) with a classical subsystem (e.g., nuclei) on a near-term quantum computer [27].
- Initialization:
- Classical System: Initialize the classical coordinates and momenta, (\overrightarrow{q}0).
- Quantum System: Prepare the initial quantum state (\left\vert \psi0 \right\rangle) on the quantum processor. This is done by generating a parameterized quantum circuit, (\hat{C}(\overrightarrow{\theta}0)), that approximates the ground state of the initial Hamiltonian (H(\overrightarrow{q}0)) using a variational quantum algorithm.
- Time Propagation Loop (e.g., Ehrenfest Dynamics): For each timestep: a. Measure Observables: Measure a set of observables ({\hat{O}^{(s)}}) from the current quantum state (\left\vert \psit \right\rangle). These are typically forces on the classical particles. b. Evolve Classical System: Use the measured expectation values to propagate the classical coordinates to the next timestep, generating (\overrightarrow{q}{t+1}). c. Update Hamiltonian: Construct the new time-dependent Hamiltonian (H(\overrightarrow{q}{t+1})). d. Evolve Quantum State: Apply a compressed time-evolution operator (e.g., via the p-VQD algorithm) to evolve the quantum state from (\left\vert \psit \right\rangle) to (\left\vert \psi_{t+1} \right\rangle), which is faithful to the new Hamiltonian.
Workflow and Pathway Visualizations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Energy and Affinity Prediction
| Item / Resource | Function & Application |
|---|---|
| PDBbind Database | A comprehensive database of protein-ligand complexes with experimentally measured binding affinities, commonly used as a benchmark for training and testing scoring functions [73]. |
| PDBbind CleanSplit | A curated version of the PDBbind training set, created by removing complexes that are structurally similar to those in the CASF test sets. It is essential for rigorously testing model generalization [73]. |
| Machine Learning Interatomic Potentials (MLIPs) | A new generation of potentials used to predict the properties of materials and molecules, including Gibbs free energy, with high computational efficiency [71] [72]. |
| Time-Dependent Variational Quantum Propagation (TDVQP) | A quantum algorithm for performing mixed quantum-classical dynamics on near-term quantum computers. It is well-suited for short-time evolution simulations [27]. |
| Reaction Network (RN) | A computational framework that uses known thermodynamic data to make experimentally informed predictions of Gibbs free energy, helping to validate and refine direct computational predictions [71]. |
Assessing the Computational Scaling and Near-Term Prospects for Quantum Advantage
Troubleshooting Common Implementation Challenges
FAQ: Addressing Researcher Questions
Q: My mixed quantum-classical dynamics simulation is experiencing rapid error accumulation. What are the primary sources of this error?
A: Error accumulation stems from multiple sources in the hybrid quantum-classical workflow [27]:
- Circuit Compression Infidelity: The algorithm approximates the time-evolved state |ψ(t+Δt)⟩ with a parameterized circuit. The difference between the true state and the compressed representation introduces state infidelity (I) at each timestep [27].
- Approximate Time Evolution: Using a first-order Trotter expansion or other NISQ-friendly time evolution methods instead of the exact propagator contributes to coherent error [27].
- Observable Measurement Noise: Noisy estimates of observables (e.g., energy gradients) from the quantum computer are fed into the classical propagation step, causing errors in the updated classical parameters and the subsequent quantum Hamiltonian [27].
- Neglected Pulay Forces: In molecular dynamics, approximating the total energy gradient by using only the Hellmann–Feynman term (ignoring Pulay forces) can reduce accuracy, though it is a common simplification for feasibility [22].
Q: For a mixed quantum-classical dynamics problem, when should I choose the Time-Dependent Variational Quantum Propagation (TDVQP) method over an approach like SHARC-TEQUILA?
A: The choice depends on your system's characteristics and computational objectives [27] [2] [22]:
- TDVQP is a more general meta-algorithm for time-dependent Hamiltonians where the quantum and classical subsystems are co-evolved in locked steps. It is suited for systems where the quantum subsystem is treated in a local diabatic basis, avoiding the need for explicit nonadiabatic coupling measurements [27].
- SHARC-TEQUILA is specifically designed for nonadiabatic molecular dynamics (NAMD) using Tully's fewest switches surface hopping. It leverages variational quantum algorithms (VQE, VQD) to compute electronic properties like excited state energies, gradients, and nonadiabatic coupling vectors on-the-fly in the energy basis. It is a strong choice for photochemical studies like photoisomerization or electronic relaxation [2] [22].
Q: The sampling overhead for Probabilistic Error Cancellation (PEC) is prohibitive for my utility-scale circuits. What can I do?
A: New software controls can dramatically reduce this overhead. Using the samplomatic package available for Qiskit, you can apply advanced techniques like propagated noise absorption and shaded lightcones. These methods allow you to target error mitigation to specific, annotated regions of your circuit, which has been shown to decrease the sampling overhead of PEC by up to 100x [74].
Troubleshooting Guide: Quantum Resource Estimation
Table 1: Resource and Error Profile of Selected Mixed Quantum-Classical Algorithms
| Algorithm | Key Computational Scaling Factors | Primary Error Sources | Recommended Mitigation Strategies |
|---|---|---|---|
| TDVQP [27] | Linear in number of timesteps, circuit parameters, and Pauli terms of observables. | Circuit compression infidelity, Trotterization error, noisy observable feedback. | Use adaptive ansatze, higher-order integrators, and increased shot counts for critical observables. |
| SHARC-TEQUILA [22] | Scales with the number of timesteps and the quantum resource requirements for VQE/VQD on each electronic state. | Accuracy of the variational quantum eigensolver/deflation, approximation of energy gradients. | Employ robust ansatze (e.g., k-UpCCGSD), leverage error mitigation, and use commuting Pauli clique grouping. |
| DECIDE [75] | Deterministic; requires integration of L²(L² - 2 + 2N) coupled differential equations (L = basis functions, N = env. DOF). | Representation of equations in an incomplete basis set. | Ensure the chosen basis (e.g., position, subsystem, adiabatic energy) is sufficiently complete for the system. |
Detailed Experimental Protocols
Protocol 1: Implementing Time-Dependent Variational Quantum Propagation (TDVQP)
This protocol outlines the steps for the TDVQP algorithm, a modular method for general mixed quantum-classical dynamics [27].
1. Initialization
- Classical Parameters: Define the initial vector of classical parameters, (\vec{q}_0) (e.g., nuclear coordinates).
- Initial Hamiltonian: Construct the initial Hamiltonian, (H0 := H(\vec{q}0)).
- Ansatz State Preparation: Select a parameterized quantum circuit ansatz, (\hat{C}(\vec{\theta})). Use a variational quantum algorithm (VQA) to prepare the initial state (|\psi0\rangle = \hat{C}(\vec{\theta}0)|0\rangle), which is an approximation to the ground or desired state of (H_0).
2. Iterative Time Propagation Loop (Repeat for each timestep)
- Observable Measurement: Measure the chosen set of observables ({\hat{O}^{(s)}t}) from the current quantum state (|\psit\rangle) to obtain expectation values ({O^{(s)}_t}). These are often energy gradients or forces.
- Classical Subsystem Evolution: Pass the measured observables to the classical integrator (e.g., Velocity Verlet) to update the classical parameters from (\vec{q}t) to (\vec{q}{t+1}).
- Quantum State Evolution:
- Update the Hamiltonian to (H{t} := H(\vec{q}{t})).
- Apply the time evolution operator to the state: (|\psi'{t+1}\rangle = \exp(-iH{t}\Delta t)|\psi_t\rangle). The physical implementation can be a first-order Trotterized circuit.
- Circuit Compression: Use a single step of the p-VQD algorithm to find new parameters (\vec{\theta}{t+1}) such that (|\psi{t+1}\rangle = \hat{C}(\vec{\theta}{t+1})|0\rangle) approximates (|\psi'{t+1}\rangle) to a desired fidelity threshold. This step is crucial for maintaining a feasible circuit depth on NISQ devices.
Software Note: This algorithm can be implemented using quantum computing frameworks like Qiskit, which now offers a C++ API for deeper integration with high-performance computing (HPC) systems [74].
Protocol 2: On-the-Fly Nonadiabatic Dynamics with SHARC-TEQUILA
This protocol details the setup for performing surface hopping dynamics using classical nuclei and quantum computer-driven electronic structure calculations [2] [22].
1. System Setup and Initialization
- Molecular System & Basis Set: Define the molecular geometry and choose an appropriate basis set (e.g., 6-31G). Perform a classical Hartree-Fock calculation to obtain one- and two-electron integrals.
- Initial Conditions: Generate initial classical nuclear coordinates and momenta, typically from a Wigner distribution. Select an initial electronic excited state for photochemical simulations.
- Ansatz Selection: Choose a variational ansatz suitable for ground and excited states. The k-UpCCGSD ansatz is recommended as a balance between accuracy and cost [22].
2. Dynamics Loop for a Single Trajectory
- Electronic Structure on QPU: For the current nuclear coordinates (R), execute the following on the quantum processing unit (QPU):
- Ground State: Run the VQE algorithm to obtain the ground state energy (E0) and wavefunction.
- Excited States: Run the Variational Quantum Deflation (VQD) or a penalty-based method to compute the energies (En) and wavefunctions for the necessary excited states, ensuring orthogonality to lower states.
- Property Calculation: Compute the required properties for dynamics on the QPU:
- Energy Gradients: Calculate the gradients of the energy with respect to nuclear coordinates (\frac{\partial E}{\partial R}). The Hellmann–Feynman term is often used as an approximation [22].
- Nonadiabatic Couplings (NACs): Calculate the derivative coupling vectors between electronic states.
- Transition Dipole Moments: Calculate the moments for property evaluation.
- Nuclear Propagation: Propagate the classical nuclei for a timestep (\Delta t) using the forces derived from the active electronic potential energy surface.
- Surface Hopping: At each step, compute the probability of hopping to another electronic state based on the NACs and the nuclear velocities. Use a stochastic algorithm to decide whether a hop occurs.
Implementation Note: This method is implemented by interfacing the molecular dynamics package SHARC with the quantum computing framework TEQUILA. It has been validated on systems like the cis–trans photoisomerization of methanimine [2] [22].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Software and Hardware for Mixed Quantum-Classical Experiments
| Item / "Reagent" | Function / Purpose | Example Platforms / Formulations |
|---|---|---|
| Quantum Software Framework | Provides tools for constructing, compiling, and executing quantum circuits. Often includes built-in implementations of VQE and other algorithms. | Qiskit (IBM), TEQUILA, TKet [74] [2] [22] |
| Classical Dynamics Package | Manages the propagation of classical degrees of freedom and integrates feedback from the quantum subsystem. | SHARC (for NAMD), Custom C++/Python Integrators [27] [2] |
| Variational Ansatz | A parameterized quantum circuit that serves as a template for representing the electronic wavefunction. Its expressibility is critical for accuracy. | k-UpCCGSD, Hardware-Efficient Ansatz (HEA), Unitary Coupled Cluster (UCC) [22] |
| Error Mitigation Suite | A collection of software techniques to reduce the impact of noise on results without requiring full quantum error correction. | Qiskit Samplomatic (for PEC overhead reduction), Zero-Noise Extrapolation (ZNE), Probabilistic Error Cancellation (PEC) [74] |
| Quantum Processing Unit (QPU) | The physical hardware that executes the quantum circuit. Performance is measured by qubit count, gate fidelity, and coherence time. | IBM Heron & Nighthawk processors, Quantinuum ion-trap systems, Atom Computing neutral-atom arrays [74] [44] |
Workflow Visualization
Conclusion
The implementation of mixed quantum-classical dynamics is progressing rapidly, driven by advances in theoretical frameworks like the exact factorization and the practical application of hybrid quantum-classical algorithms. While challenges such as managing electronic decoherence, phase evolution, and algorithmic errors remain active research areas, the successful application of these methods to real-world drug discovery problems—from prodrug activation to covalent inhibitor design—signals a transformative shift in computational biochemistry. Future directions will focus on developing more robust error correction techniques, refining the integration of quantum computing into biological simulation pipelines, and ultimately achieving a demonstrable quantum advantage that can reshape the landscape of biomedical research and clinical drug development.
References
- 1. Unifying Decoherence and Phase Evolution in Mixed ... [https://arxiv.org/html/2511.05794v1]
- 2. SHARC meets TEQUILA: mixed quantum-classical dynamics ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc04987j]
- 3. Quantum and Mixed Quantum Classical Dynamics in ... [https://www.cecam.org/workshop-details/quantum-and-mixed-quantum-classical-dynamics-in-photochemistry-1416]
- 4. How quantum computing is changing drug development ... [https://www.weforum.org/stories/2025/01/quantum-computing-drug-development/]
- 5. Quantum computing in life sciences and drug discovery [https://www.mckinsey.com/industries/life-sciences/our-insights/the-quantum-revolution-in-pharma-faster-smarter-and-more-precise]
- 6. Quantum mechanics implementation in drug-design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5587087/]
- 7. Decoherence in Quantum Computing: Causes, Effects, Fixes [https://www.spinquanta.com/news-detail/decoherence-in-quantum-computing-everything-you-need-to-know]
- 8. Quantum decoherence [https://en.wikipedia.org/wiki/Quantum_decoherence]
- 9. Unifying Decoherence and Phase Evolution in Mixed ... [https://arxiv.org/abs/2511.05794]
- 10. Quantum Computing for Dummies: A Complete Beginner's ... [https://www.spinquanta.com/news-detail/quantum-computing-for-dummies-a-complete-beginner-s-guide-to-the-future-of-technology]
- 11. The Potential Role of Quantum Computing in Biomedicine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12096140/]
- 12. Analysis of divergent dynamics of exactly factorized ... [https://arxiv.org/html/2510.07460]
- 13. Exact Factorization Adventures: A Promising Approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9267945/]
- 14. Different flavors of exact-factorization-based mixed ... [https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp03464j]
- 15. Nonadiabatic Excited-State Dynamics with Machine Learning [https://pmc.ncbi.nlm.nih.gov/articles/PMC6174422/]
- 16. Optimization of mixed quantum-classical dynamics [https://www.sciencedirect.com/science/article/abs/pii/S0301010408004783]
- 17. New computational chemistry techniques accelerate the ... [https://nse.mit.edu/computational-chemistry-predicting-molecules-materials/]
- 18. Advancing Universal Deep Learning for Electronic ... [https://arxiv.org/html/2509.19877v2]
- 19. Mixed Quantum-Classical Methods for Polaron Spectral ... [https://arxiv.org/html/2505.13365v1]
- 20. Basics of Surface Hopping in Mixed Quantum/Classical ... [https://www.sciencedirect.com/science/article/pii/S0021999199962873]
- 21. Quantum Computing 2025: Qubit Counts ... [https://learningbreeze.com/physics/the-quantum-reality-a-deep-dive-into-the-state-of-quantum-computing-in-2025/]
- 22. SHARC meets TEQUILA: mixed quantum-classical dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11653199/]
- 23. SHARC meets TEQUILA: Mixed Quantum-Classical ... [https://chemrxiv.org/engage/chemrxiv/article-details/66a35167c9c6a5c07a00cd88]
- 24. Practical techniques for high-precision measurements on ... [https://www.nature.com/articles/s41534-025-01066-1]
- 25. Performance Evaluation of Variational Quantum ... [https://arxiv.org/html/2503.24045v2]
- 26. Quantum Computing 2025: Beyond Hype to Real ... [https://medium.com/@daveshpandey/quantum-computing-2025-beyond-hype-to-real-breakthroughs-e45132b20e0c]
- 27. Mixed quantum-classical dynamics for near term ... [https://www.nature.com/articles/s42005-023-01451-2]
- 28. Complex Fluid Models of Mixed Quantum–Classical ... [https://link.springer.com/article/10.1007/s00332-024-10044-4]
- 29. What Is Quantum Computing? Full Beginner Guide 2025 [https://www.spinquanta.com/news-detail/what-is-quantum-computing-and-how-does-it-work-expert-explained]
- 30. Your Complete Guide to Quantum Computing Sessions [https://aws.amazon.com/blogs/quantum-computing/aws-reinvent-2025-your-complete-guide-to-quantum-computing-sessions/]
- 31. Quantum algorithm for protein-ligand docking sites ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12228596/]
- 32. Protein Solvation Pipeline [https://open-quantum-institute.cern/protein-solvation-pipeline/]
- 33. Quantum computing makes waves in drug discovery [https://www.stjude.org/research/progress/2025/quantum-computing-makes-waves-in-drug-discovery.html]
- 34. A general framework for active space embedding methods ... [https://www.nature.com/articles/s41524-024-01477-2]
- 35. A general framework for active space embedding methods [https://arxiv.org/html/2404.18737v2]
- 36. New data analysis method accelerates design of safer covalent ... [https://www.news-medical.net/news/20250930/New-data-analysis-method-accelerates-design-of-safer-covalent-inhibitors.aspx]
- 37. A hybrid quantum computing pipeline for real world drug discovery [https://www.nature.com/articles/s41598-024-67897-8?error=cookies_not_supported&code=e960c607-f402-4a32-b5da-d6820d3fa57f]
- 38. Discovery of orally active and serine-targeting covalent ... inhibitors [https://pubmed.ncbi.nlm.nih.gov/41132852/]
- 39. A hybrid quantum computing pipeline for real world drug ... [https://www.nature.com/articles/s41598-024-67897-8]
- 40. Adaptive quantum dynamics with the time-dependent ... [https://arxiv.org/html/2506.08575v4]
- 41. Can Error Mitigation Improve Trainability of Noisy ... [https://quantum-journal.org/papers/q-2024-03-14-1287/]
- 42. Variational optimization for quantum problems using deep ... [https://www.nature.com/articles/s42005-025-02261-4]
- 43. Quantum computing breakthrough could slash pesky errors ... [https://www.livescience.com/technology/computing/this-moves-the-timeline-forward-significantly-quantum-computing-breakthrough-could-slash-pesky-errors-by-up-to-100-times]
- 44. Quantum Computing Industry Trends 2025: A Year of ... [https://www.spinquanta.com/news-detail/quantum-computing-industry-trends-2025-breakthrough-milestones-commercial-transition]
- 45. On the consistency of measurement protocols for quantum ... [https://arxiv.org/html/2502.12905v2]
- 46. Quantum feedback [https://en.wikipedia.org/wiki/Quantum_feedback]
- 47. Real-Time Two-Axis Control of Spin Qubits [https://www.quantum-machines.co/blog/feedback-driven-quantum-stabilization-two-axis-real-time-control-of-spin-qubits/]
- 48. An initialization strategy for addressing barren plateaus in ... [https://quantum-journal.org/papers/q-2019-12-09-214/]
- 49. Can Classical Initialization Help Variational Quantum ... [https://arxiv.org/html/2508.18497v1]
- 50. Quantum Strategies to Overcome Classical Multiplexing ... [https://arxiv.org/html/2510.06099v2]
- 51. Evaluating the performance of classical, quantum, and ... [https://www.nature.com/articles/s41598-025-22950-y]
- 52. A coherent approach to quantum-classical optimization [https://www.nature.com/articles/s42005-025-02111-3]
- 53. Nonadiabatic molecular dynamics on quantum computers [https://www.degruyterbrill.com/document/doi/10.1515/pac-2025-0599/html?lang=en&srsltid=AfmBOop4So2drrwm1jt7icf-i2B3hS2Ro48oqkkBnOq0QvRjVNChRzTK]
- 54. Reliable Optimization Under Noise in Quantum Variational ... [https://arxiv.org/html/2511.08289v1]
- 55. Fast gradient-free optimization of excitations in variational ... [https://www.nature.com/articles/s42005-025-02375-9]
- 56. Constraints on Quantum-Advantage Experiments Due to Noise [https://link.aps.org/doi/10.1103/Physics.18.174]
- 57. Isomerization and electronic relaxation of azobenzene after ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326308001629]
- 58. Control over molecular motion using the cis–trans ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3458724/]
- 59. Photoisomerization Dynamics of Azo-Escitalopram Using ... [https://chemrxiv.org/engage/chemrxiv/article-details/670d8a47cec5d6c142269a6b]
- 60. Photoisomerization mechanism of iminoguanidinium ... [https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp02747g]
- 61. Computational Comparison of Chemical and Isotopic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8154570/]
- 62. Hybrid Quantum-Classical Methods - UChicago Voices [https://voices.uchicago.edu/triplehelix/2025/05/20/hybrid-quantum-classical-methods-the-future-of-quantum-computing-and-ai/]
- 63. Temporal quantum interference in many-body ... [https://arxiv.org/html/2511.09633v1]
- 64. Landau-Zener-Stückelberg spectroscopy of a fluxonium ... [https://arxiv.org/html/2504.21691v1]
- 65. A Quantum Shield Law for Coherence and Tunnelling [https://chemrxiv.org/engage/chemrxiv/article-details/68e782a2dfd0d042d198ca99]
- 66. Nonadiabatic Wave Packet Dynamics [https://www.emergentmind.com/topics/nonadiabatic-wave-packet-dynamics]
- 67. Surface hopping [https://en.wikipedia.org/wiki/Surface_hopping]
- 68. 9.8.6 Fewest-Switches Surface Hopping [https://manual.q-chem.com/5.3/sect_fssh.html]
- 69. beyond Nonadiabatic Couplings for... Surface Hopping Dynamics [https://pubmed.ncbi.nlm.nih.gov/29439572/]
- 70. Simulations of photoinduced processes with the exact ... [https://pubs.rsc.org/en/content/articlehtml/2024/cp/d4cp02489c]
- 71. Performance assessment of high-throughput Gibbs free ... [https://www.sciencedirect.com/science/article/pii/S0927025625001132]
- 72. Performance assessment of high-throughput Gibbs free ... [https://orbit.dtu.dk/en/publications/performance-assessment-of-high-throughput-gibbs-free-energy-predi/]
- 73. Resolving data bias improves generalization in binding ... [https://www.nature.com/articles/s42256-025-01124-5]
- 74. Scaling for quantum advantage and beyond [https://www.ibm.com/quantum/blog/qdc-2025]
- 75. A Deterministic Mixed Quantum-Classical Dynamics ... [https://www.mdpi.com/2076-3417/12/14/7022]